
基于GEM展示技术内含肽介导的犬α干扰素快速纯化系统的建立
2019-07-06 06:11:12仝晴晴乔绪稳张元鹏郑其升牛家强侯继波
中国动物传染病学报 2019年3期
仝晴晴,杨 利,乔绪稳,陈 瑾,张元鹏,郑其升,牛家强,侯继波
(1. 西藏农牧学院动物科学学院,西藏 860000;2. 江苏省农业科学院 国家兽用生物制品工程技术研究中心,江苏 210014)
蛋白纯化是疫苗制备中十分重要的一步,纯化标签是可用于纯化多种靶蛋白的工具。如何通过廉价高效的方法对目的蛋白进行快速的纯化是研究中急需被攻破的课题[1]。内含肽自我剪切的发现为快速高效纯化目的蛋白奠定了基础[2]。内含肽是前体未成熟蛋白的一段具有自我剪接功能的多肽链。1990 年,第一个内含肽酿酒酵母(Saccharomyces cerevisiae)VMA1被发现[3],随后内含肽在蛋白质纯化、蛋白质连接、环肽制备、蛋白标记以及生物传感器等方面被广泛应用[4-5]。112 aa的N片段和35 aa的C片段组成的工程化的NpuDnaE内含肽能够在没有N-末端剪切的情况下快速催化C-末端剪切[6-8]。NpuDnaE突变体表现出硫代依赖性的C末端快速剪切[9-10],将锚钩蛋白(protein anchor)定位到内含肽N片段的N末端,可避免N片段和C片段之间相互作用的潜在空间位阻,使得超快速剪切成为可能[1,11]。
GEM(gram positive enhancer matrix)颗粒是乳酸菌经热酸处理,去除细菌原有蛋白质和核酸等大分子物质后获得的细胞壁肽聚糖骨架[12],具有特异性非共价结合细菌细胞壁水解酶C端结构域蛋白质(protein anchor,PA)的特性[13]。GEM与含3个LysMs的PA(PA3)的结合活性高于含2个LysMs的PA(PA2)[14]。因此,与PA3连接的内含肽及目的蛋白,可以高密度地展示在GEM颗粒表面形成沉淀[15],仅需低速离心即可纯化目的蛋白。
天然干扰素制备工艺复杂、产量低、成本高,而利用大肠杆菌表达系统表达外源蛋白操作简单,成本低廉、可控性好[16]。本研究利用大肠杆菌表达系统开发了断裂型NpuDnaE内含肽介导的基于GEM展示平台的蛋白纯化方法,其中N和C外显子被锚钩蛋白(PA3)和目的蛋白(CaIFN-α)取代,利用GEM颗粒与锚钩蛋白的特异性结合得到较为纯净的蛋白GEM-PA3-NC1A,再利用断裂型NpuDnaE内含肽的N端和C端结合获得融合蛋白GEM-PA3-NC1AC*-CaIFN-α,最后利用内含肽的硫代剪切特性获得纯净的目的蛋白CaIFN-α[17](图1)。本研究将断裂型NpuDnaE内含肽与GEM展示系统结合,建立了一种简洁高效的蛋白纯化方法,并以犬α干扰素(CaIFN-α)为目的蛋白验证了该方法的可行性,同时确定了蛋白纯化的最佳条件。

图1 蛋白表达与纯化示意图Fig.1 Schematic of protein expression and purification
1 材料和方法
1.1 质粒、细胞和病毒内含肽N端和锚钩蛋白的重组表达质粒pQZ-NC1A-PA3以及内含肽C端表达质粒pET-C*由本实验室保存;含有犬α干扰素基因的重组质粒pMD18-CaIFN-α由江苏省农科院兽医研究所王永山研究员提供;大肠杆菌感受态细胞DH5α和BL21 由本实验室保存;犬肾细胞(madindarby canine kidney,MDCK)、水疱性口炎病毒(Vesicular stomatitis virus,VSV,107.0TCID50/mL)由本实验室保存。
1.2 主要试剂限制性内切酶NheI、EcoR I、T4 DNA连接酶购自TaKaRa 公司;DTT和EDTA购自Sigma公司;琼脂粉、蛋白胨、氯化钠、氯化锌等常规试剂为国产分析纯;所有缓冲液使用ddH2O制备。
1.3 引物设计根据 GenBank 中公布的CaIFN-α基因序列进行优化后,在编码成熟蛋白基因的两端设计引物,为方便基因操作在5'和3'端分别加入NheI、EcoR I酶切位点。上下游引物序列:5'-GCTAGCTGCCATCTGCCGGATACC-3',5'-GAATTC GCTTATTTGCGGCGACGAATACGT TCC-3',由英潍捷基(上海)贸易有限公司合成。
1.4 CaIFN-α基因的克隆与表达载体的构建通过PCR从pMD18-CaIFN-α质粒中扩增获得CaIFN-α基因,克隆至pMD18-T载体,送TaKaRa 公司进行测序以确定序列的正确性。PCR扩增体系为50 μL:1 μL pMD18-CaIFN-α质粒作为模板,25 μL master mix(TaKaRa Japan)和1 μL正向和反向引物。PCR循环参数:95℃预变性5 min,95℃变性30 s,56℃退火30 s,72℃延伸30 s;一次最终变性在72℃延伸5 min。将pMD18-T-CaIFN-α和pET-C*质粒分别用NheI和EcoR I双酶切,电泳回收酶切产物,用T4 DNA连接酶于16℃连接过夜。连接产物转化DH5a感受态细胞,挑选阳性克隆菌,提取质粒后经NheI和EcoR I双酶切鉴定。
1.5 重组菌的诱导表达及蛋白提取将pQZ-NC1A-PA3和pET-C*-CaIFN-α重组表达载体分别转化至BL21感受态细胞中,挑选阳性克隆菌于 LB 培养液(含适宜抗生素)中,37℃过夜培养,次日将5 mL培养物转移至400 mL LB培养基中,并在37℃下生长直至OD600约为0.6。加入阿拉伯糖和IPTG(0.2 mmol/L)在15℃诱导表达24 h后,4℃、8000×g离心10 min,收获细胞,在蒸馏水中洗涤2次。然后将pQZNC1A-PA3和pET-C*-CaIFN-α分别重悬于缓冲液A(0.5 mol/LNaCl、10 mmol/LTris-HCl,pH8.0)和缓冲液B(0.5 mol/LNaCl、50 mmol/LNaPOi,pH 6.0)中。超声破碎细胞,4℃、12 000×g离心10 min,收集可溶性裂解物,用0.45 μm滤器过滤,并保存于-80℃。利用SDS-PAGE鉴定蛋白的表达及蛋白可溶性。
1.6 CaIFN-α蛋白的纯化
1.6.1 NC1A-PA3与GEM颗粒的结合及鉴定 取出10 U制备好的GEM颗粒,离心后用20 mL含有NC1A-PA3可溶性裂解物的缓冲液A重悬,室温下缓慢转动孵育1 h。离心收集沉淀,将沉淀用等体积缓冲液B洗涤3次,最后1次洗涤缓冲液中含有0.5 mmol/L ZnCl2。SDSPAGE鉴定结合结果。
1.6.2 内含肽的结合及鉴定 将洗涤后的沉淀加至等体积的含C*-CaIFN-α可溶性裂解物的缓冲液B中重悬,17℃缓慢转动孵育3 h。离心收集沉淀,用等体积的缓冲液A洗涤3次,SDS-PAGE鉴定结合结果。
1.6.3 内含肽剪切及内毒素的去除 将沉淀用4 mL含有DTT或EDTA的缓冲液A重悬,4℃缓慢转动孵育过夜。剪切后离心,收集上清和沉淀,进行SDSPAGE分析。通过3 kDa超滤离心柱(Amicon Ultra,Millipore,Billerica,MA)浓缩纯化的蛋白质,经NanoDrop 1000(Thermo Fisher Scientific)测定OD280值。
1.7 微量细胞病变抑制法为测定上述纯化得到的目的蛋白的抗病毒活性,先将犬肾细胞 (MDCK) 接种于96孔板,5%CO2、37℃条件下培养至单层,加入倍比稀释的干扰素作用18 h后,每孔再加入100 TCID50的VSV,同时设置阴性对照组,48~72 h后观察细胞病变结果。
2 结果
2.1 重组表达载体的鉴定将PMD18-T-CaIFN-α和pET-C*质粒分别用NheI和EcoR I酶切,将回收纯化的大小片段连接后转化DH5a感受态细胞,提取的质粒经酶切电泳鉴定分离出约 520 bp的小片段即为CaIFN-α,证实在pET-C*中插入了CaIFN-α基因,所得质粒命名为PET-C*-CaIFN-α。
2.2 重组质粒的表达及可溶性鉴定收集表达后的菌体,按1.5中方法进行裂菌处理,取裂解液的上清和沉淀进行 SDS-PAGE电泳分析。结果显示,pQZNC1A-PA3表达产物的相对分子量约为37 kDa,pETC*-CaIFN-α表达产物的相对分子量约为26 kDa,两种菌体蛋白表达总量较大,上清中均存在目的蛋白,杂蛋白较多(图2)。

图2 重组 pQZ-NC1A-PA3和pET-C*-CaIFN-α表达产物的SDS-PAGE分析Fig.2 SDS-PAGE of recombinant pQZ-NC1A-PA3 and pETC*-CaIFN-α expression in E.coli
2.3 CaIFN-α蛋白的纯化
2.3.1 NC1A-PA3与GEM颗粒的结合及鉴定 利用GEM颗粒与锚钩蛋白非特异性结合的特性,将NC1A-PA3蛋白吸附于GEM颗粒上,形成GEM-PA3-NC1A沉淀(图3)。因总蛋白量较多,结合过程中其他杂蛋白也会吸附在GEM颗粒表面,但不影响纯化结果。
2.3.2 内含肽的结合及鉴定 利用内含肽的结合特性,将C*-CaIFN-α与GEM-PA3-NC1A沉淀结合形成GEM-PA3-NC1A-C*-CaIFN-α沉淀(图4)。结合过程中可适当浓缩以提高纯化蛋白的浓度。
2.3.3 内含肽剪切及蛋白纯化 将内含肽结合后沉淀用含DTT或EDTA的缓冲液A重悬,利用DTT和EDTA对内含肽反式剪切的催化作用,将目的蛋白还原为可溶性的CaIFN-α蛋白,离心弃去的沉淀为GEM-PA3-NC1A-C*及其他吸附在GEM颗粒上的杂蛋白。经SDS-PAGE电泳后染色,结果如图5所示,纯化后缓冲液上清中杂蛋白很少,纯化效果良好,且EDTA的剪切效果较DTT更好。
2.4 CaIFN-α蛋白的活性测定应用MDCK-VSV系统微量细胞病变抑制法测定纯化后的CaIFN-α蛋白活性。结果显示接种VSV 48 h后,纯化的犬α干扰素的抗病毒比活性为3.0×106U/mL。
3 讨论
分别表达内含肽的两个片段,让其在体外相互识别结合并完成剪接反应,可以解决提前断裂的问题,且其反应速率并未受到影响[18-19]。内含肽改造是通过突变剪接结构模块中的保守氨基酸,尤其是疏水氨基酸,使剪接反应只能完成一端的断裂[20-21]。有研究通过突变内含肽内部的2个氨基酸为Cys,使其与首位氨基酸Cys之间形成二硫键,从而抑制断裂反应提前进行[22]。Asp118 是阻止NpuDnaE C 端断裂反应的关键因素[10],将Asp118 突变为Gly后,可解除N端突变对C端断裂反应的抑制。天然断裂型NpuDnaE 内含肽的断裂反应速率较快,改造后的NpuDnaE 内含肽不仅剪切速率提高,而且避免了提前断裂的发生。Zn2+通过抑制蛋白质剪接过程中的电荷传递影响内含肽剪接反应,使剪切反应无法顺利进行[23]。虽然DTT与Zn2+的结合能力强于内含肽与Zn2+的结合,但比金属螯合剂(EDTA)的结合能力弱,因此可通过EDTA将Zn2+从内含肽中去除以恢复剪接反应[24]。
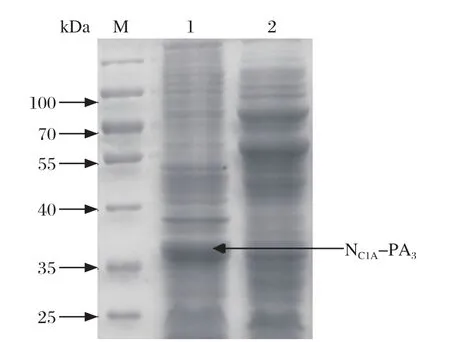
图3 GEM颗粒与NC1A-PA3结合的SDS-PAGE分析Fig.3 SDS-PAGE of NC1A-PA3 incubation with GEM

图5 GEM-PA3-NC1A-C*-CaIFN-α融合蛋白剪切的SDSPAGE分析Fig.5 SDS-PAGE of cleavage of GEM-PA3-NC1A-C*-CaIFN-α fusion protein.
GEM表面展示系统已应用于许多疫苗研发中。Bosma等[12]利用该系统研制了疟原虫表面抗原MSA2的亚单位疫苗。人肺炎链球菌粘膜疫苗[25]、鼠疫LcrV亚单位疫苗等[26],以及多价苗的研制都利用了GEM表面展示系统。本实验室利用GEM表面展示系统浓缩纯化了O型口蹄疫病毒灭活抗原[27]和猪繁殖与呼吸综合征病毒[15]。
本研究建立了一种基于GEM展示平台,由断裂型NpuDnaE内含肽介导的蛋白纯化方法,简便易操作:利用GEM颗粒沉淀可溶的目的蛋白,不需要对包涵体进行变性复性等复杂操作,蛋白损失较小;利用内含肽剪切不需要昂贵的酶和几丁质柱子等额外成本,仅依靠还原剂DTT或EDTA,通过离心即可获取较为纯净的目的蛋白;不需要层析介质,避免了层析介质与配基蛋白片段结合不稳定而造成的蛋白脱落。同时,IN与IC在体外结合作用更强,可以减少目标蛋白中出现前体蛋白[28]。结果表明,将非层析标签与断裂内含肽系统相结合构建的纯化系统为纯化目的蛋白提供了一种新思路,成本低,易于操作与放大,具有广阔的应用前景。
猜你喜欢
中国科技纵横(2021年24期)2021-03-02 06:42:52
中成药(2018年8期)2018-08-29 01:28:34
中成药(2018年6期)2018-07-11 03:01:12
中国继续医学教育(2015年6期)2016-01-07 07:38:50
哈尔滨医药(2015年6期)2015-12-01 03:58:19
中国医疗美容(2015年2期)2015-07-19 10:11:59
医学研究杂志(2015年9期)2015-07-01 17:28:24
中国当代医药(2015年9期)2015-03-01 02:02:13
河南医学研究(2014年4期)2014-02-27 14:52:28
山西大同大学学报(自然科学版)(2013年5期)2013-09-13 10:44:14
