
拟南芥突变基因位置的Linux大数据分析
2019-06-18 13:36:24甘秋云
唐山师范学院学报 2019年3期
甘秋云
拟南芥突变基因位置的Linux大数据分析
甘秋云
(福州理工学院 文理学院,福建 福州 350014)
以拟南芥为实例,通过野生型和突变型杂交,对二代群体DNA进行深度测序,获取海量DNA数据。以SNP为分子标记,利用生物信息学方法对测序数据进行单核苷酸多态性(SNP)检测。通过置换测验对基因组区段内的等位基因频率进行差异显著分析,并利用生物统计学方法对具有显著性差异的数据进行显著性检验,预估拟南芥的突变位点的位置在1号染色体的末端位置,范围为2 853 000~2 898 000。
基因突变;基因定位;等位基因;高通量测序
突变是发生于DNA水平上的一种永久性变化,可能发生碱基对组成或排列顺序的改变。对突变的研究不仅为育种工作提供必要的实验材料,对科学研究和生产也有重要意义[1,2,3]。本文结合计算机算法,生物统计学和数学的计算方法,对测序数据进行分析、处理,预测突变基因在染色体上的位置。
1 数据来源与技术方法
1.1 数据
以拟南芥为研究对象,将群体野生型和突变型进行杂交,对二代群体DNA进行深度测序,获得29 264 012条序列读段,长度为76 bp。过滤、筛选后获得27 215 530条,平均长度为76 bp的序列读段。由拟南芥数据库TAIR[4]下载拟南芥参考基因序列,利用SOAPaligner软件,将上述读段与参考基因组序列进行对比,检测单核苷酸多态性(SNP),最终获得全基因组范围内SNP位点[4]。上述全基因组序列读段作为定位突变基因的分析对象。
1.2 样品百分数△d计算
采用不同的移动窗口,固定步长,分别在野生型和突变型两个DNA池中,计算出相应窗口下的SNP位点的碱基个数y1与参考碱基一致的读段的个数2,SNP位点的测序深度1和2。然后,通过公式(1)计算相应的基因频率(野生型标记为“1”,突变型标记为“2”)。
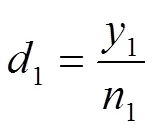
针对某些样本容量较小的区间,在容量基础加上一个常数(=50)。
基因频率相应写为
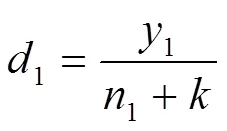
两个样本基因频率的差值为
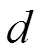
通过△值的变化曲线图,预测突变位点的位置。
1.3 差异显著分析
对野生型和突变型的2个DNA池的样品进行1 000次随机分组,得到1 000组随机的样本文件。利用计算机算法计算1 000次随机实验中位于同一区间下的最大值,作为差异显著性分析的数据来源。
采用Aspin-Welch方法对预估的显著区间数据进行显著性检验,求出在总体显著水平为5%时的的阈值P。若P值小于0.05,则认为当前数据差异是显著的,找到差异显著的区间在染色体上的位置,可初步判断该位置可能存在一个突变位点或目标基因[4]。
该检验中的临界值由t表查出,自由度由
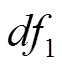
确定。其中,
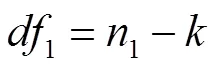
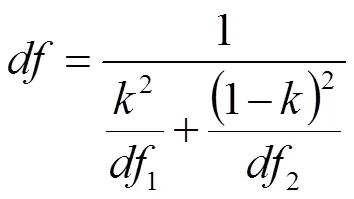
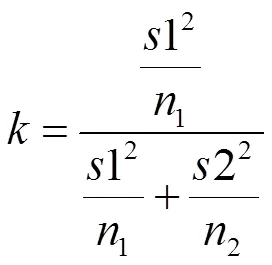
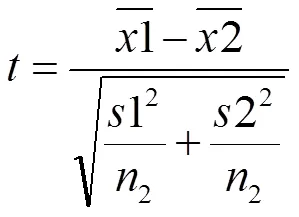
以10 kb为移动步长,分别设置100 kb、200 kb、300 kb、400 kb的移动窗口,利用公式(2)分别计算野生型和突变型两个样本在不同染色体对应位点的基因频率。根据计算结果,以染色体位置为横坐标,△值大小为纵坐标,绘制全基因组范围内的△值变化图。
2 结果与分析
2.1 样本百分比统计
图1是1号染色体在移动窗口为400 kb时的值曲线图。从图中明显看到,在1号染色体末端出现了高峰区间。
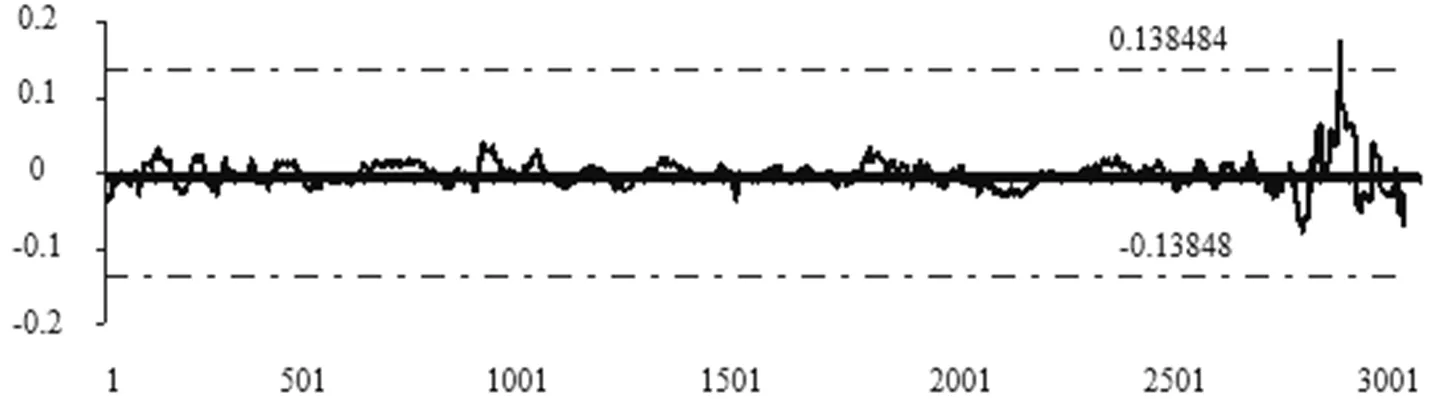
图1 拟南芥野生型与突变型1号染色体在移动窗口400 kb下△d值曲线图
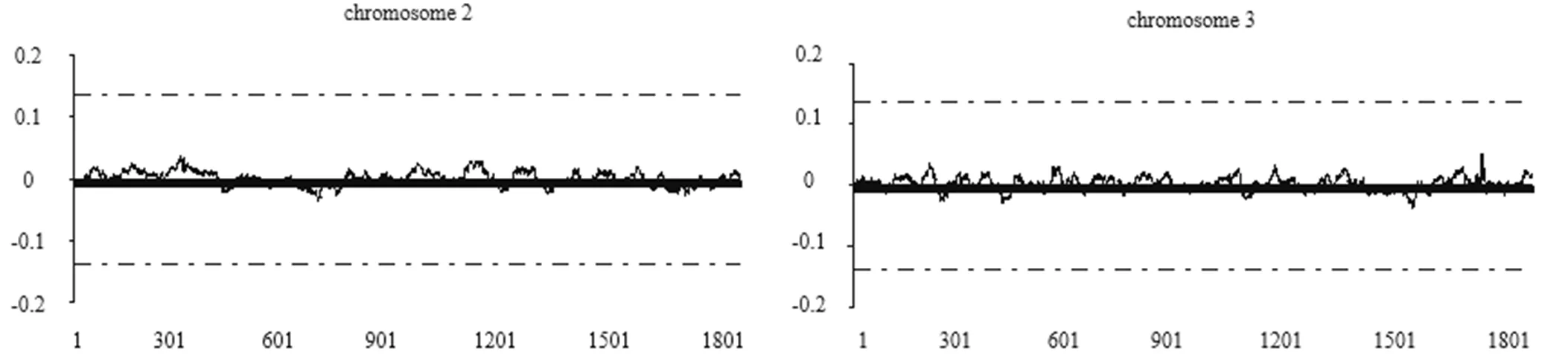
图2分别是2、3、4、5号染色体在移动窗口为400 kb时值曲线图。从图中可见,2至5号染色体的值变化较小,曲线整体走势较平缓。
2.2 检测突变位点
根据1 000组随机实验得到的值,绘制出对应的分布图,发现其分布情况满足正态分布特点。
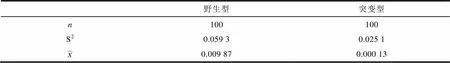
表1 野生型和突变型显著性分析数据
抽取1号染色体末端位置上的100个△数据样本,分析结果见表1。
方差齐性检验结果为
H0:σ1=σ2;HA:σ1≠σ2;α=0.05
在显著水平0.05的前提下,
F=75.923,F0.05=1.25,F≠F0.05,
方差不具齐性。
Aspin-Welch检验结果为
H0:μ1=μ2;HA:μ1≠μ2;α=0.05
将数据带入公式(4)、(6),得到的值为101.61,t值为3.03。利用=101.61时的t临界值使用线性内插法求出t=1.984。
由于t>t0.05,即p<0.05,所以在野生型和突变型两个DNA池样本在1号染色体末端位置出现的高峰区间具有显著性差异,可以初步预测当前位置突变位点的位置。
图3是1号染色体的值曲线图。
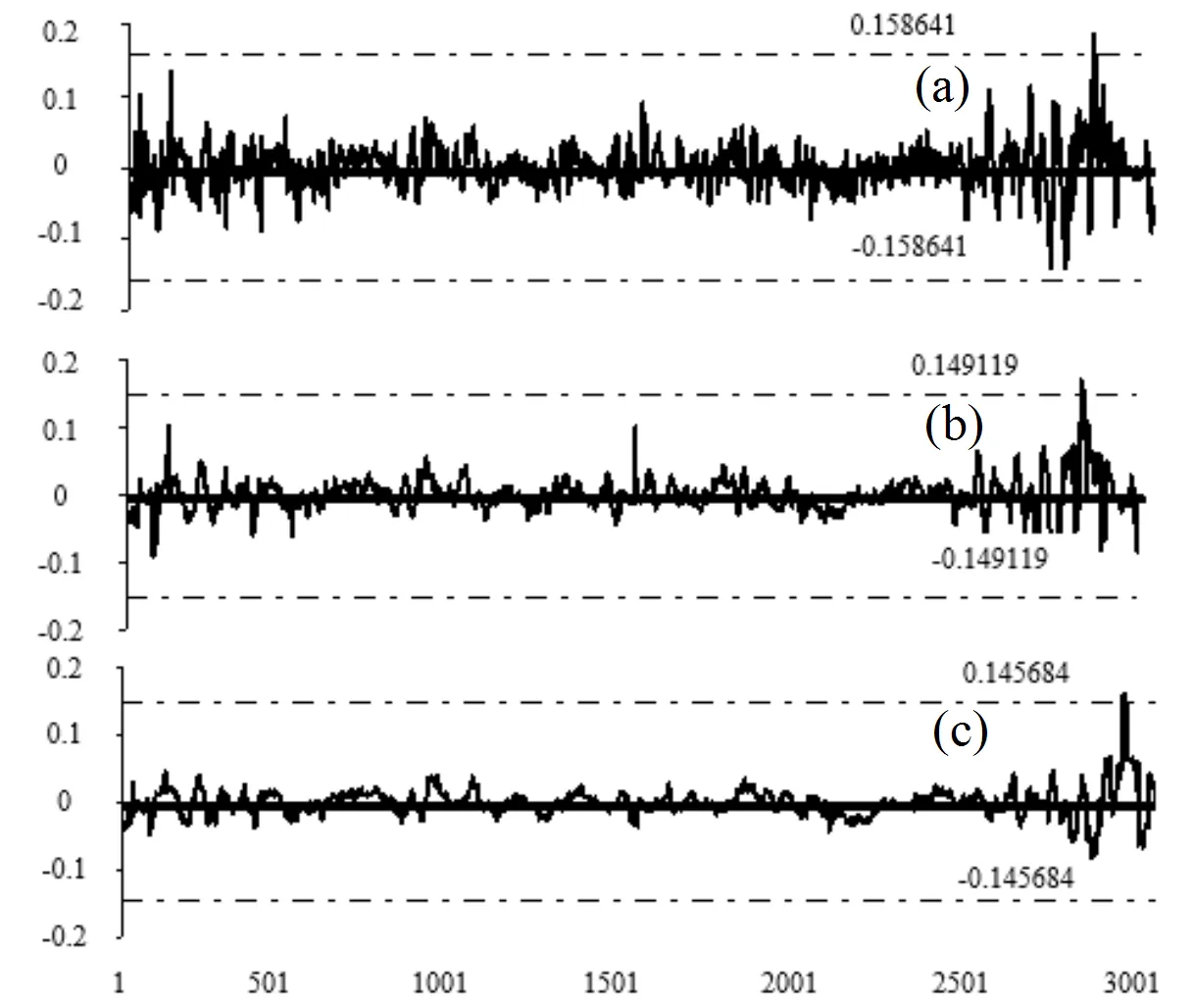
(a)100 kb;(b)200 kb;(c)300 kb
从图3可以发现,1号染色体在不同的移动窗口下都出现了高峰区间。随着移动窗口的不同,样本高峰值不同,在移动窗口为100 kb达到最大,为0.188 953。在200 kb、300 kb、400 kb的移动窗口下高峰值平均为0.17。但1号染色体的高峰区间均位于染色体末端。
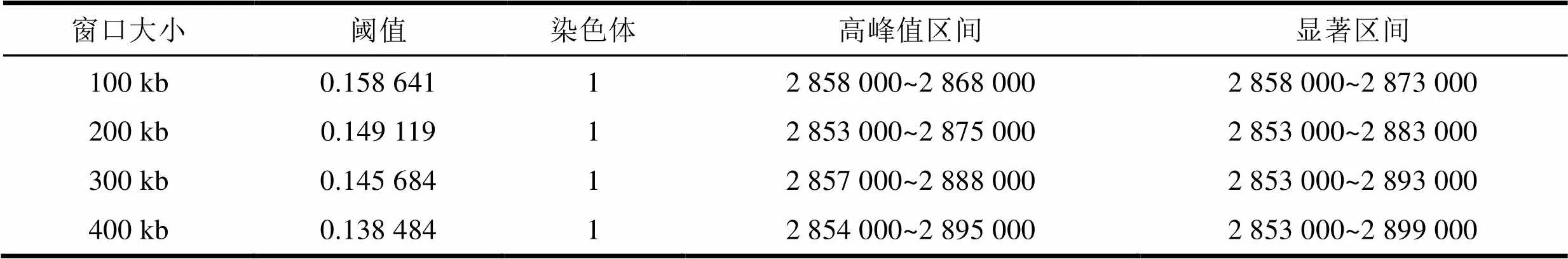
表2 1 000组随机显著实验统计拟南芥染色体显著区间(显著水平=0.05)
通过4次不同移动窗口的1 000组随机实验,得到如表2所示的位于1号染色体上的显著区间的分布情况。从表2可以判断,突变位点位于1号染色体的位置区间范围为2 853 000 ~ 2 898 000。
3 结论
利用深度测序获取拟南芥野生型和突变型杂交二代群体DNA数据。以SNP为分子标记,综合运用计算机、数学、生物统计学等方法对SNP位点进行处理、分析,计算等位基因频率并进行差异显著分析。预测出了目标基因或突变位点的位置在1号染色体的末端,突变位点出现的区间位置范围为2 853 000~2 898 000。
[1] 张玲.基于全基因组测序及外显子组测序的食管癌相关基因筛选及功能鉴定[D].太原:山西医科大学,2015:1.
[2] 李维,刘若余,冯艳青,李思,杜雪琴,谢海强,肖超能,林家栋.家兔UCP3基因SNP多态性及生物信息学分析[J].基因组学与应用生物学,2015,(10):2127-2133.
[3] 王媛,韩如意,苏玉贞,孙丽,张连民,王晨.人CITED4基因及蛋白的生物信息学分析[J].生物技术,2016,26(6): 566-573.
[4] 甘秋云.利用深度测序定位拟南芥突变基因[D].福州:福建农林大学,2011:14-18.
Mapping of Arabidopsis Mutant Gene Based on Large Data Analysis of Linux
GAN Qiu-yun
(School of Arts and Sciences, Fuzhou Institute of Technology, Fuzhou 350014, China)
In this paper, Arabidopsis thaliana is taken as an example to obtain massive DNA data by deep sequencing of DNA from the second generation population through wild-type and mutant hybridization. Single nucleotide polymorphism (SNP) detection of sequencing data was conducted by using bioinformatics methods and using SNP as a molecular marker. Significant differences in the frequency of alleles within the genome segment were analyzed by a displacement test. The data of significant differences were statistically tested by biostatistical methods to estimate the location of the mutation site of Arabidopsis thaliana.
gene mutation; gene mapping; allele; high throughput sequencing
TP399
A
1009-9115(2019)03-0060-04
10.3969/j.issn.1009-9115.2019.03.017
2017-09-27
2019-03-04
甘秋云(1986-),女,福建宁德人,硕士,讲师,研究方向为计算机应用,数据挖掘,生物信息学。
(责任编辑、校对:李春香)
猜你喜欢
中学生天地(A版)(2023年1期)2023-02-17 00:33:04
科学之谜(2019年3期)2019-03-28 10:29:44
科学之谜(2018年8期)2018-09-29 11:06:46
生命科学研究(2018年1期)2018-05-29 01:12:47
上海农业学报(2017年3期)2017-04-10 12:39:14
山东农业工程学院学报(2016年6期)2016-12-01 05:38:19
恋爱婚姻家庭·养生版(2016年9期)2016-09-07 11:25:01
中央民族大学学报(自然科学版)(2015年2期)2015-06-09 08:45:16
安徽医药(2014年9期)2014-03-20 13:14:09
吉林大学学报(医学版)(2014年2期)2014-02-27 06:48:05
