
选择性雌激素受体下调剂研究进展
2019-06-06 07:40钮嘉辉王小伟尤启冬
药学进展 2019年4期
钮嘉辉 ,王小伟,尤启冬
(1.中国药科大学药学院,江苏 南京210009;2.南京圣和药业股份有限公司研发中心,江苏 南京210038)
乳腺癌是一种发生在乳腺上皮组织的恶性肿瘤。世界卫生组织国际癌症研究中心公布的2012年全球肿瘤流行病统计数据(GLOBOCAN 2012)显示,乳腺癌是全球女性发病率最高的癌症,致死率居第6位[1]。在我国,虽然乳腺癌发病率与致死率显著低于世界平均水平,但是每年乳腺癌新发病例数已达28万左右[2]。
雌激素受体(estrogen receptor,ER)作为乳腺癌分子分型的重要判断依据,其在乳腺癌组织中表达与否很大程度上决定了患者对于乳腺癌内分泌治疗的反应以及预后。据临床统计,约60% ~ 75%的乳腺癌患者其ER表达呈阳性[3]。而ER信号通路在ER阳性乳腺癌癌细胞的增殖、转移等方面都起到非常重要的作用。由此可见,ER对于ER阳性乳腺癌治疗来说是一个重要靶点。
选择性雌激素受体下调剂(selective estrogen receptor down-regulators,SERDs),作为新一代以ER为靶点的ER阳性乳腺癌内分泌治疗药物,以其独有的作用机制以及临床表现吸引了许多药企的目光。国内外多家药企近年来都开展了SERDs的研发。
1 雌激素受体
1.1 雌激素受体结构
ER是属于甾体超家族的一类核受体[4],包括ERα和ERβ。这2个亚型结构相似,从N端到C端依次为A、B、C、D、E、F共6个功能结构域(见图1)。A/B区含有不依赖配体的转录活性区域AF-1(activation function-1),通过与转录因子、辅激活因子等相互作用传递信号,使得靶基因被激活[5]。C区域为DNA结合域(DNA binding domain,DBD)。D/E/F区域为配体结合域(ligand binding domain,LBD),主要功能为调整ER与配体结合,受体二聚化以及下游基因激活。ER配体结合域还包含了一个配体依赖的转录活性区域AF-2。其中AF-2区域重要组成部分螺旋结构-12在SERDs与ER结合、ER降解方面起到重要作用。
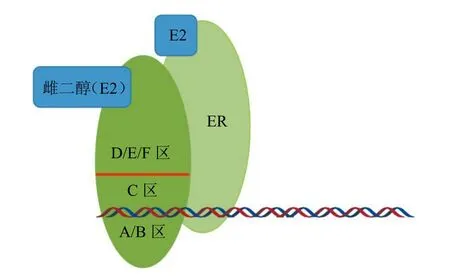
图1 雌激素受体结构Figure 1 The structure of estrogen receptor
ERα/ERβ 2个亚型在AF-1、AF-2区域的同源性都较低,分别为15%和53%[6],表明2种亚型之间存在一定的差异。这也导致2种亚型拥有特异性的配体并具有不同的生物学功能。其中ERα对乳腺癌中雌激素诱导基因起到主导性的调控作用[7]。
1.2 雌激素受体信号通路
ER信号通路分为核信号通路(见图2中通路A、B、C)以及膜信号通路(见图2中通路D)[8],不仅调节着人类正常的生理活动,在乳腺癌发生及发展中也具有重要作用。
在未激活状态下,ER与热休克蛋白90(heat shock protein 90,Hsp90)结合形成寡聚体复合物[8]。经典的配体依赖信号通路中,内源性雌激素与ER结合,ER构象发生改变,Hsp90脱落。接着ER以同源或异源二聚体的形式与靶基因上雌激素反应元件(estrogen response element,ERE)结合,在相关辅因子协同作用下,调节下游基因的转录。如下调miR-140的表达,促使乳腺癌干细胞启动[9]。在不依赖雌激素的情况下,ER通过生长因子激活的相关胞内信号通路作用,使得自身磷酸化[10]。磷酸化活化的ER与ERE结合,对下游靶基因的转录进行调控,促进乳腺癌癌细胞增殖。ER还可以通过与靶基因上游转录因子间的相互作用,参与不含ERE基因的转录调控。以原癌基因Cyclin D1为例,ER能与其编码基因CCND1启动子区域刺激蛋白1结合,导致过度转录[11]。除了上述占主导作用的核信号通路,一部分镶嵌于细胞膜上的ER介导的膜信号通路,如丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)/细胞外信号调节蛋白激酶(extracellular signal regulating protein kinase,ERK)等通过改变细胞内部对应蛋白的功能,会导致细胞过度增殖或抑制细胞凋亡,对正常生理活动造成一定影响,对乳腺癌的发展也起到一定作用。
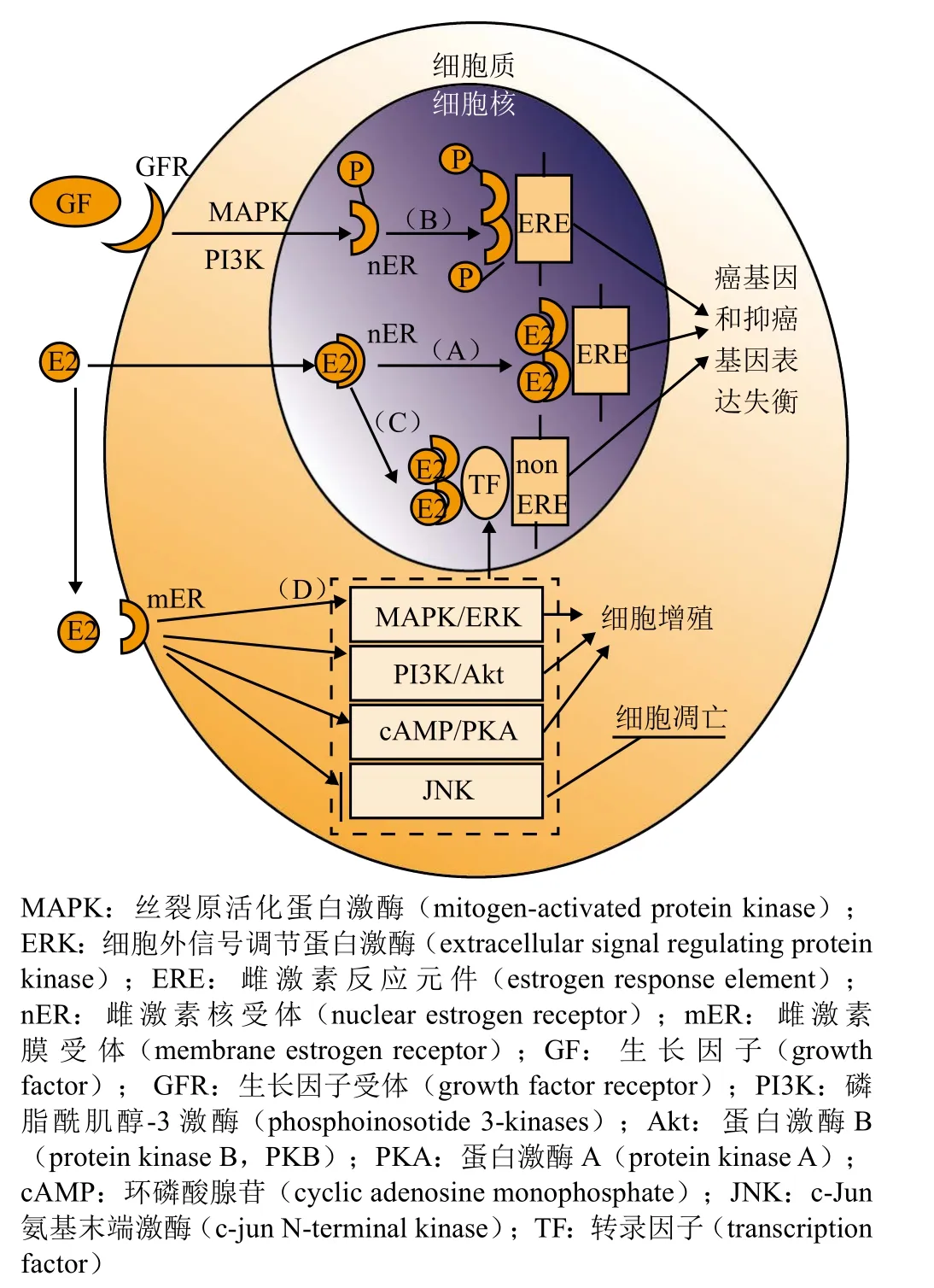
图2 雌激素受体信号通路Figure 2 The estrogen receptor signaling pathway
2 以雌激素受体为靶点的乳腺癌治疗药物
目前,以ER为靶点的乳腺癌内分泌治疗药物根据作用机制不同可分为3类:选择性雌激素受体调节剂(selective estrogen receptor modulators,SERMs)、SERDs以及选择性雌激素受体共价拮抗剂(selective estrogen receptor covalent antagonists,SERCAs)。
其中SERMs通过拮抗内源性雌激素与ER的结合,令ER复合物处于拮抗构型,使得靶基因的转录衰减,细胞周期被阻断在G1期,从而表现出抑制乳腺癌癌细胞增殖的作用[12]。作为SERMs代表药物的他莫昔芬(tamoxifen,1),上世纪70年代开始被用于乳腺癌辅助内分泌治疗,时至今日在乳腺癌等肿瘤治疗与预防中仍发挥着作用。但是他莫昔芬存在如下几个显著缺点:1)患者在持续性治疗中会产生耐药现象;2)在子宫中表现出激动剂作用,患者罹患子宫内膜癌风险明显上升[13];3)会导致血栓栓塞风险增高[14]。这些都限制了其在乳腺癌治疗中更广泛的应用。而以雷洛昔芬(raloxifene,2)、拉索昔芬(lasofoxifene,3)为代表的第2、3代SERMs,虽然解决了他莫昔芬使子宫内膜癌患病风险上升的缺陷,但是在乳腺癌临床治疗方面并未表现出明显优势。目前,第2、3代SERMs在乳腺癌领域的临床应用主要体现在乳腺癌高危人群的化学预防。
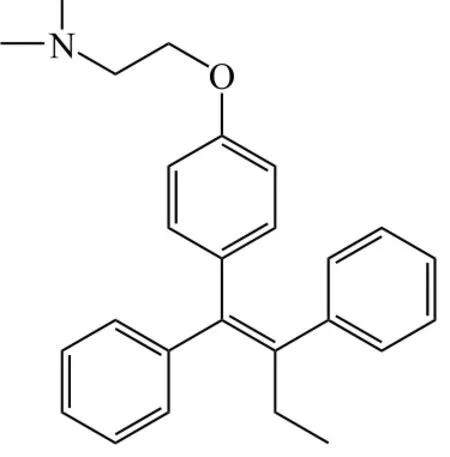
1
2
SERDs在拮抗ER的基础上,通过与ER结合降低其稳定性,促使其被胞内蛋白酶体降解而显著降低ER水平。ER水平的下调会对ER信号通路产生全面抑制,从而达到抑制癌细胞增殖的效果。相较于SERMs仅仅影响经典配体依赖的ER信号通路,SERDs能够对ER信号通路产生更加全面的抑制。由于作用机制的不同,也规避了例如耐药以及子宫内膜癌患病风险上升等缺陷。
3 选择性雌激素受体下调剂
3.1 氟维司群
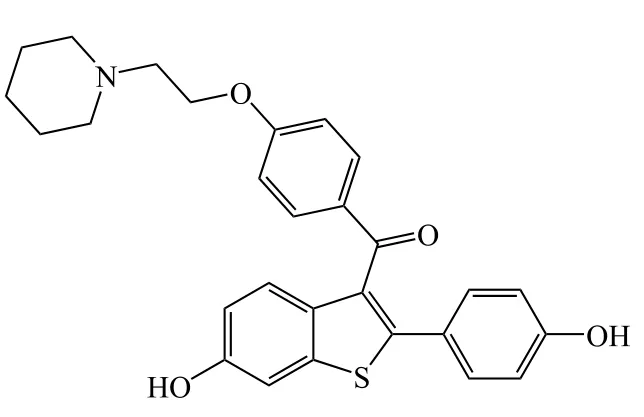
氟维司群(fulvestrant,4)是由阿斯利康(Astra Zeneca)开发的首个SERDs,2002年经美国FDA批准上市,也是目前唯一一个已上市的SERDs药物。
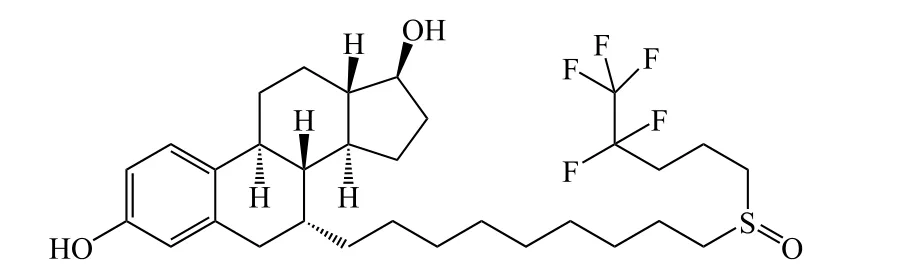
4
最初阿斯利康打算开发一款ER完全拮抗剂,但是在细胞实验中却发现氟维司群能够下调ERα水平,因此对其下调ERα的机制进行了探索。Yeh等[15]发现氟维司群能够在不影响胞内ERα mRNA水平情况下显著降低ERα水平。同时利用蛋白酶体抑制剂MG132预处理细胞能够逆转这一现象。可见氟维司群是通过蛋白酶体降解ER的方式下调胞内ER水平。
深入研究显示:氟维司群与ERα结合后,侧链从结合口袋中伸出,使得ERα蛋白结构中的螺旋结构-12定位受到干扰,ERα疏水性表面暴露增大,稳定性降低,更易受到蛋白酶体降解。如将螺旋结构-12突变,则对氟维司群介导的ER降解作用表现出抵抗性[16],进一步证明了螺旋结构-12在该过程中的重要性。
因而,氟维司群通过与ER结合降低其稳定性,促使其被胞内蛋白酶体降解而显著降低ER水平。但是氟维司群也会通过拮抗内源性雌激素与ER结合,阻断ER二聚化,限制其核位移而阻断靶基因的转录。氟维司群既拮抗又降解的作用机制,从2个角度对ER信号通路产生抑制,达到抑制乳腺癌癌细胞增殖的作用。
临床上氟维司群用于治疗ER阳性转移型乳腺癌,给药剂量经历了每月250 mg到每月500 mg的转变。2002年FDA批准的氟维司群给药剂量为每月250 mg。临床结果显示:该给药剂量对于ER阳性转移型乳腺癌治疗有效,但是相较于其他内分泌治疗药物(如他莫昔芬)在疾病进展时间、临床获益率等方面并未表现出明显优势[17];并且250 mg给药方案的药物稳态浓度在治疗开始后3 ~ 6个月才能到达,因此使疾病早期复发的风险增加[18]。较多因素都表明氟维司群临床用药剂量仍待进一步研究。
由于氟维司群的ER降解能力以及疗效在早期临床试验50、125、250 mg剂量组中表现出剂量依赖性[19],因此研究人员参考他莫昔芬曾进行过的高剂量给药实验,将氟维司群给药剂量增加到500 mg进行评估。根据药动学模型预测,500 mg氟维司群给药能够更快达到更高的血药浓度(见图3[19])。
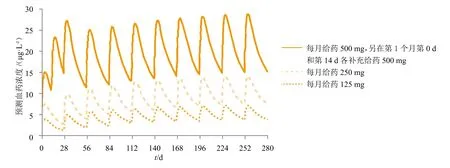
图3 药动学模型模拟500 mg氟维司群血药浓度曲线图Figure 3 Blood concentration curve of 500 mg fulvestrant simulated by pharmacokinetic model
多项临床试验结果也显示氟维司群每月500 mg给药方案表现均优于每月250 mg(见表1),且耐受性良好[20]。鉴于此,FDA于2010年再次批准氟维司群每月500 mg给药方案用于绝经后晚期ER阳性乳腺癌患者的治疗。
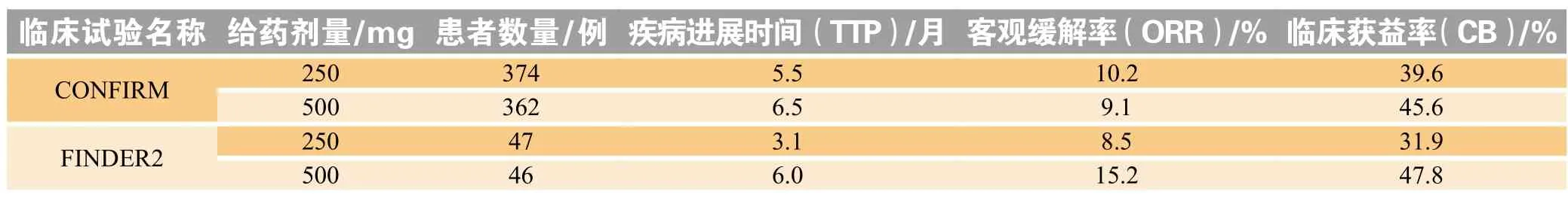
表1 氟维司群500和250 mg在临床试验中的相关数据Table 1 Related data of fulvestrant 500 and 250 mg in clinical trials
3.2 AZD9496及其衍生物
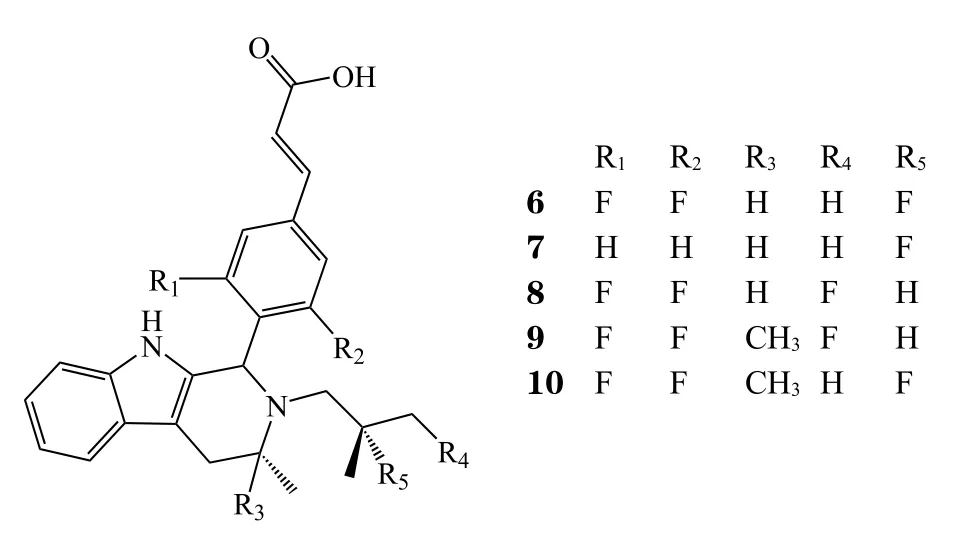
虽然氟维司群的给药方案得到了改进,但其给药剂量及给药方式(肌内注射)的限制依旧对其临床应用带来阻碍。即使是最新批准的氟维司群每月500 mg给药方案,基于18F-氟雌二醇的正电子发射型计算机断层显像(FES-PET)显示其对ER的阻断程度仍低于75%[21],这也导致了其临床疗效的下降。因此,阿斯利康继续研发SERDs,以期得到能够快速达到更高血药浓度、临床表现更佳的口服SERDs药物。他莫昔芬活性代谢产物4-OH他莫昔芬与氟维司群中的苯酚结构均与雌二醇A环结构相似,化合物与ER结合的能力虽得到增强,但是苯酚结构会导致较高的清除率和较低的生物利用度。鉴于此,阿斯利康利用高通量筛选,重新选定了不含苯酚结构的苗头化合物(5)进行修饰,得到一系列化合物(6 ~ 10)[22]。这一系列化合物在体外都表现出不错的ERα结合能力和下调ER水平的能力。
5
其中AZD9496(6)以其优秀的ERα降解能力(ERα降解IC50为0.14 nmol · L-1)从这些化合物中脱颖而出。其构效关系分析表明:1)新母核吲哚环上NH通过与Leu346的羧基形成氢键,产生相互作用;2)吲哚并四氢吡啶N原子邻位甲基模仿他莫昔芬的乙基部位,占据Phe-404/425亲脂性口袋,提升ER结合能力;3)吲哚并四氢吡啶N原子取代侧链上的2个甲基填充了Leu384/525亲脂性口袋[22];4)丙烯酸侧链与Asp351通过羧酸间相互作用共定位于螺旋结构-12区域,导致螺旋结构-12位置改变,增大了ERα疏水表面的暴露,使得ERα稳定性下降,更易被降解。其中丙烯酸侧链的存在对于提高SERDs对ERα的降解能力至关重要[23](见图4[24])。
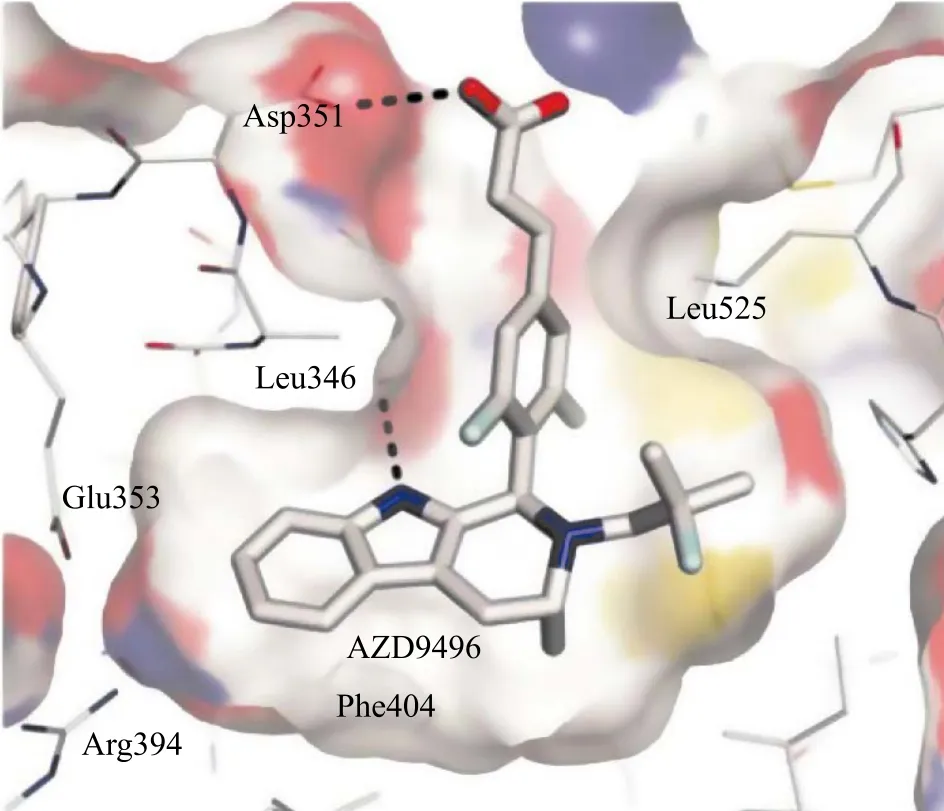
图4 AZD9496与雌激素受体α配体结合域复合物晶体结构Figure 4 Crystal structure of the complex between AZD9496 and the ligand binding domain of ERα
随后,阿斯利康以氟维司群作为对照,对AZD9496体外ERα结合、ERα降解,体内ERα降解等多项能力再次进行评估(见表2)[24]。在体内ERα降解方面,AZD9496与氟维司群的效能相当。AZD9496为5 mg ·kg-1时,AZD9496的体内抑瘤作用优于对照组氟维司群(每只小鼠1周给药3次,每次给药5 mg),肿瘤生长抑制率达75%。
AZD9496与氟维司群在体外ERα结合、ERα降解等方面无明显差异,而AZD9496体内肿瘤抑制率高于氟维司群,结合氟维司群在临床及动物模型中表现出较低的生物利用度[24],推测可能是MCF-7异种移植瘤模型中游离药物浓度的高低导致了肿瘤抑制作用存在区别。这也提示着研发人员,提高SERDs药物的血药浓度,对于其在体内发挥降解ERα,抑制癌细胞增殖作用意义重大。故AZD9496在生物利用度等方面较好的表现(见表3)为其后续临床试验的推进奠定了基础[22]。
AZD9496的Ⅰ期临床试验分别于2014年、2015年在美国和英国开展。一项有45例患者参与的Ⅰ期临床试验显示:AZD9496具有较好的耐受性及安全性,且对接受过治疗的ER阳性、人表皮生长因子受体(human epidermal growth factor receptor 2,Her-2) 阴性的晚期乳腺癌患者具有延长疾病稳定时间的作用[25]。
除了阿斯利康等国外大型药企,国内许多药企也敏锐地意识到SERDs在乳腺癌治疗领域所具有的潜在优势,紧跟脚步立项研发。例如,浙江海正药业以AZD9496作为模板,通过结构修饰以期获得活性更佳的SERDs。其结构修饰通式如图5所示,并对这些化合物申请了专利(专利号:WO2018001232)[26]。
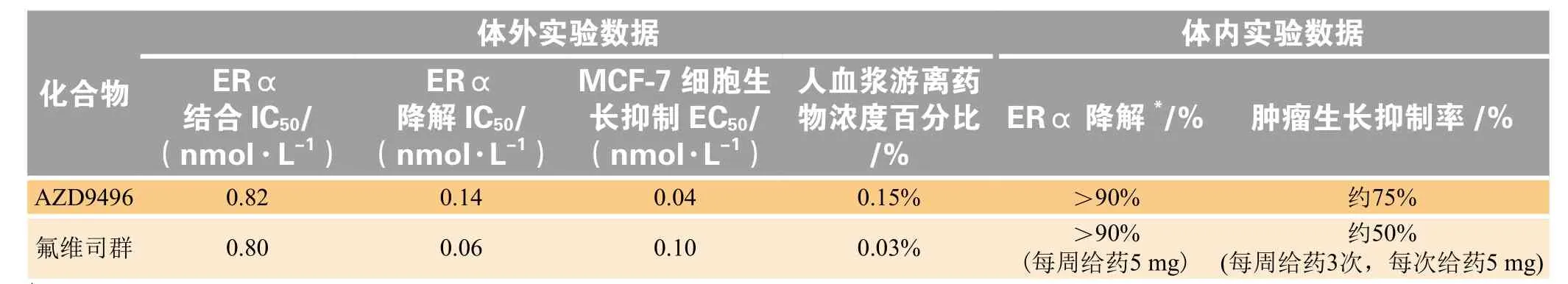
表2 AZD9496与氟维司群体内外相关数据比较Table 2 Comparison of in vitro and in vivo data between AZD9496 and fulvestrant
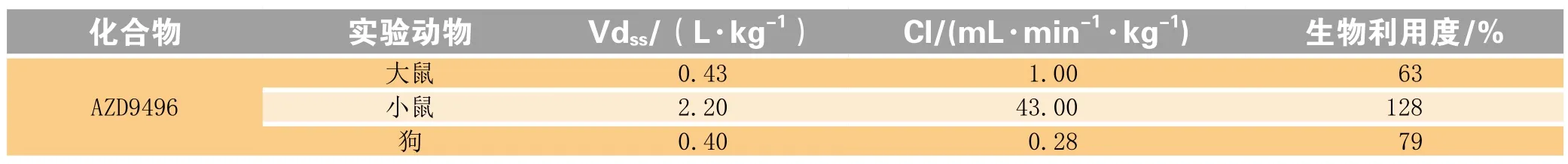
表3 AZD9496在动物体内实验中的药动学参数Table 3 Pharmacokinetic parameters of AZD9496 in animal experiments
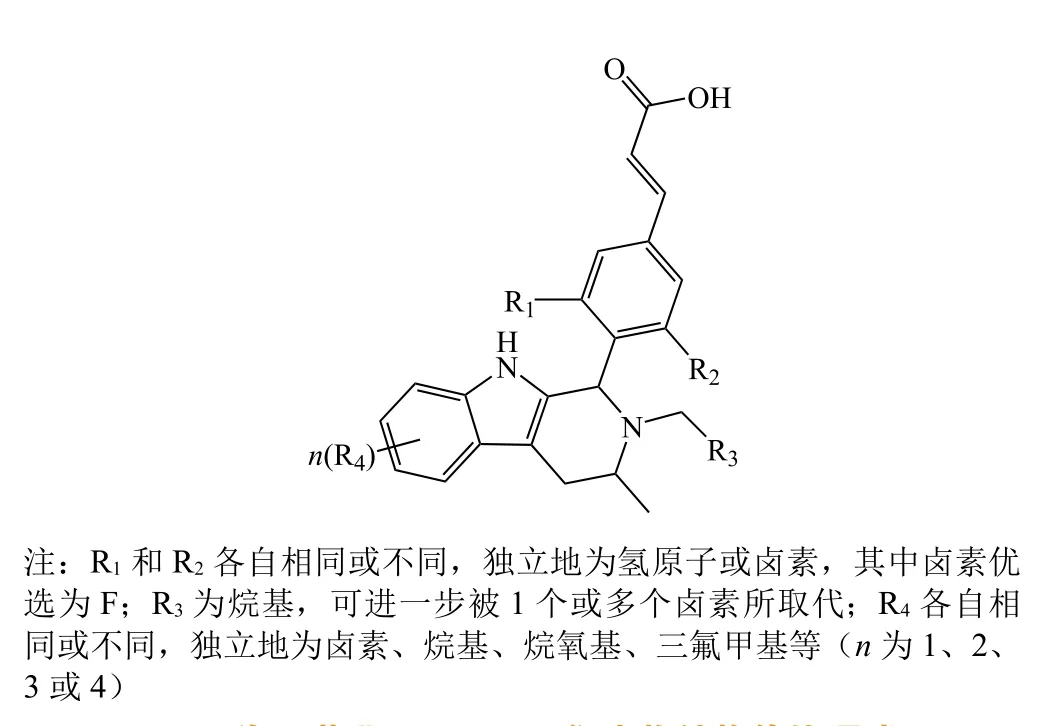
图5 海正药业AZD9496衍生物结构修饰通式Figure 5 The general modification structure of AZD9496 derivatives patented by Haizheng
上述专利保护中的化合物在ER结合以及ERα下调方面,IC50都达到了纳摩尔水平。根据披露的体外活性数据,化合物11在专利包含的化合物中表现最佳,10 mg · kg-1时产生的肿瘤生长抑制率在给药第21 d达80.4%,略优于同剂量的AZD9496(76.5%),具有与其他公司化合物竞争的能力。
恒瑞医药在SERDs药物开发上同样以AZD9496为模板,但该公司对AZD9496的结构改动比较大,用苯环、萘环等取代基替换母核中的吲哚部分(结构修饰通式1见图6),得到了一系列化合物并进行了专利保护(专利号:WO2016202161)[27]。
在图6中A区域改动得到的十几个系列化合物中,苯环系列整体表现较佳。恒瑞医药对苯环系列的再改造包括进行并环、杂环取代等,得到近百个化合物。其中带吡唑取代基的苯环系列中有多个化合物(12 ~ 14)都表现出较好的体外活性(见表4)[27]。
为了改善代谢问题,恒瑞医药针对R2部位进行了结构修饰,结构通式如图7所示[28]。修饰后的化合物在ER结合能力、ERα降解能力以及MCF-7细胞增殖抑制方面表现也更加出色。基于ERα降解、MCF-7细胞增殖抑制等表现,恒瑞医药从中选取了一个化合物,命名为SHR9594,作为候选化合物继续推进研发。由于第2次结构修饰的专利并未公开所有化合物的体外活性数据,所以并不清楚SHR9594的具体结构。目前SHR9594的临床研究申请已向国家药品监督管理局(NMPA)递交,Ⅰ期临床试验也正处于准备阶段。
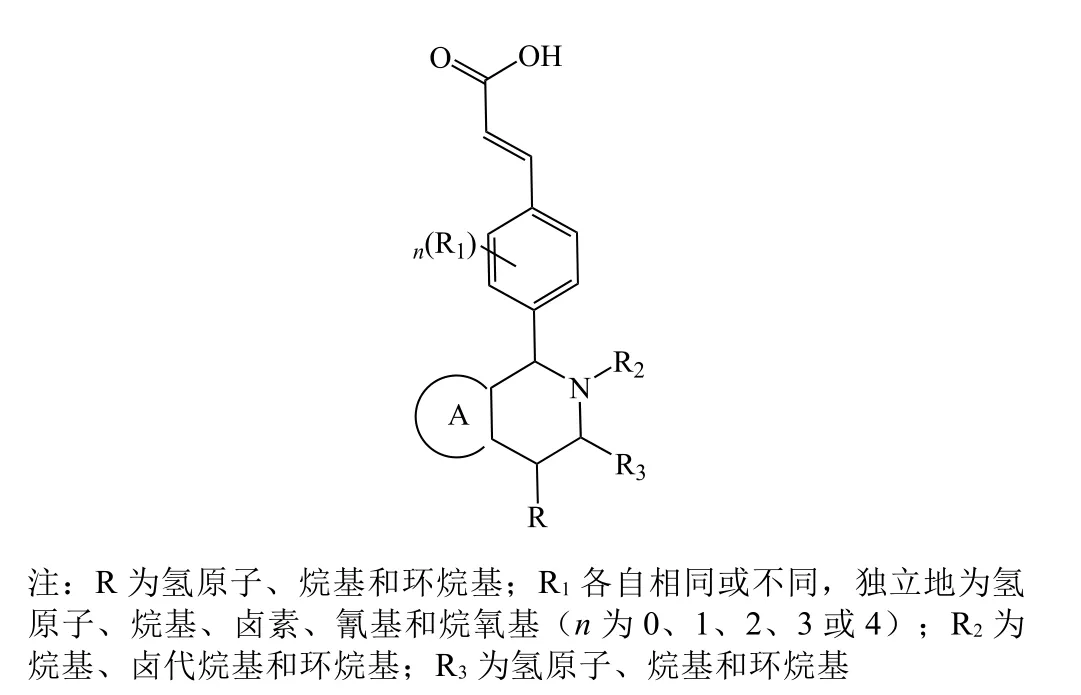
图6 恒瑞医药AZD9496衍生物结构修饰通式1Figure 6 The general modification structure I of AZD9496 derivatives patented by Hengrui
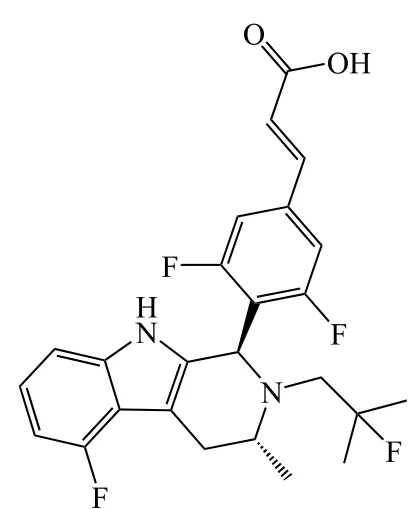
11
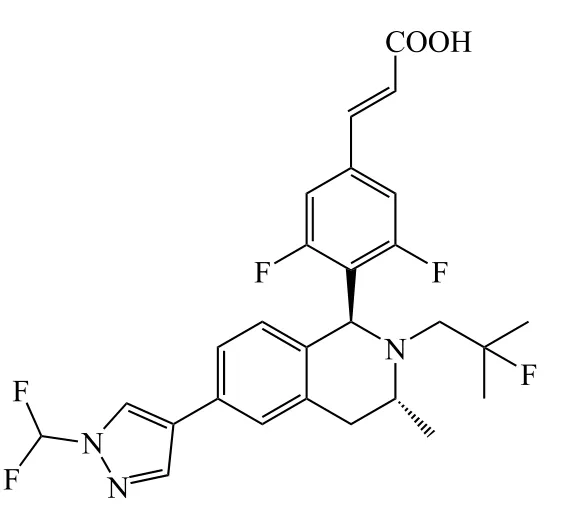
12
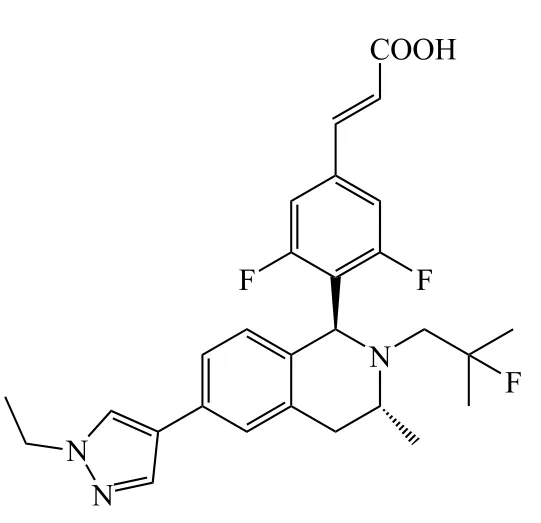
13
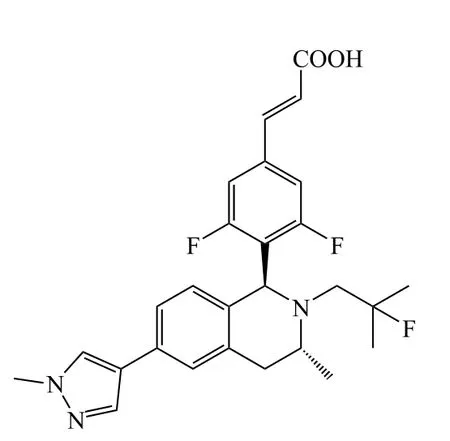
14
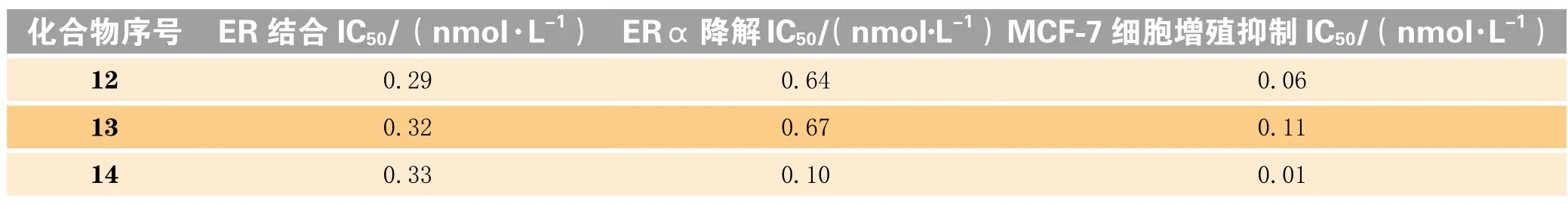
表4 WO2016202161中的部分化合物体外活性数据Table 4 In vitro activity data of some compounds in WO2016202161
3.3 GDC-0810及其衍生物/GDC-0927
研究发现,葛兰素威康(Glaxo Wellcome)公司开发的他莫昔芬类似物GW-5638(15)在某些组织中(如乳腺)表现出SERDs的性质[29]。基因泰克(Genentech)公司选取该化合物作为结构修饰对象,将改造区域划分为A、B、C等3个区域,得到多个系列的化合物(16 ~20)。对A区域进行的结构修饰发现,相当多的芳杂环取代基取代在A区域后,化合物体外ERα降解EC50均达到微摩尔甚至纳摩尔水平[30]。当A区域取代基为吲哚基、吲唑基时,化合物ERα降解及MCF-7细胞增殖抑制表现较优(见表5),其中,吲唑基取代的一系列化合物在小鼠动物模型中经口给药时都表现出较好的生物利用度及药动学性质[30]。因此在同时开发多个系列化合物时,基因泰克公司将目光重点放在了吲哚及吲唑基取代系列上,含有吲唑基取代的GDC-0810(20)为代表化合物。
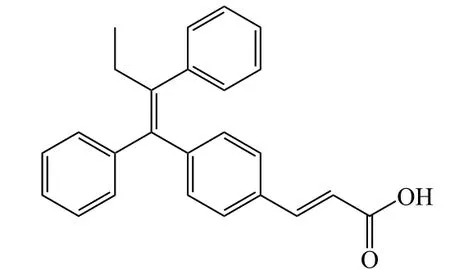
15
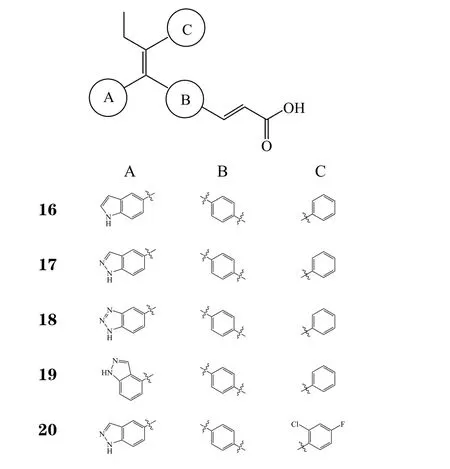
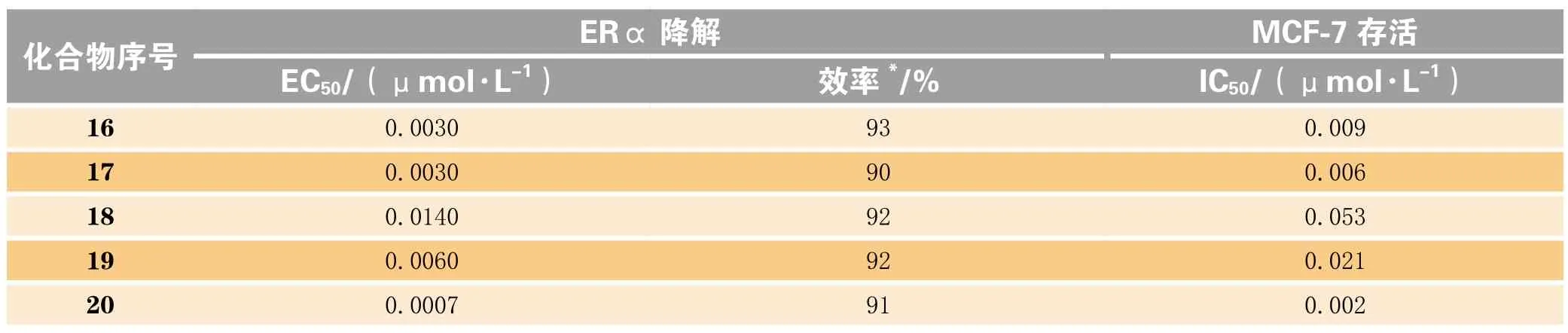
表5 基因泰克公司结构修饰部分化合物体外数据Table 5 In vitro activity data of some modified compounds patented by Genentech
采用他莫昔芬敏感、他莫昔芬耐药2个肿瘤模型评估化合物GDC-0810体内抑瘤效果时,结果显示100 mg ·kg-1GDC-0810表现出的抑瘤能力优于氟维司群(200 mg · kg-1,每周3次);其中在他莫昔芬耐药模型中,GDC-0810抑制肿瘤体积的效果是氟维司群的6倍[31]。在针对患有ER阳性晚期或转移性乳腺癌绝经后女性患者的Ⅰ期临床试验中,FES-PET成像显示,GDC-0810对ER的阻断能力强于氟维司群,90%的实验对象表现出90%以上的ER阻断[32]。
基因泰克公司以化合物GDC-0810为基础,再次进行了结构修饰。用1-氟乙基、1-氯乙基、异丙基、环丙基、环丁基等取代基取代后得到数个系列的化合物(21 ~25)。当取代基为环丁基时,化合物的体外活性更佳,有发展潜力(见表6)[33]。
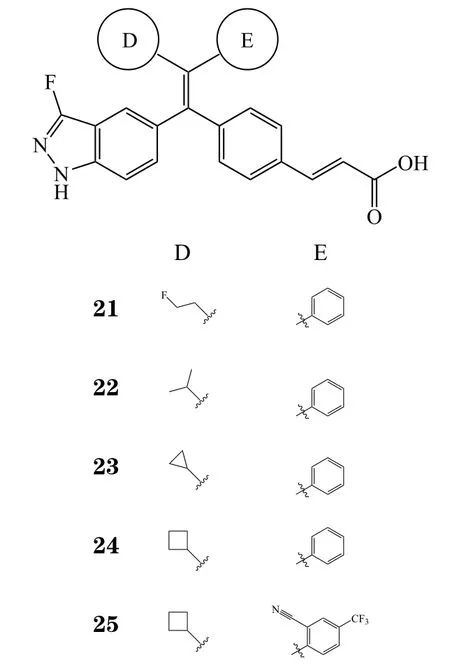
其中化合物25在D区域为环丁基、环丙基等空间体积相近取代基的系列分子中(21 ~ 25)体外活性表现最佳。在他莫昔芬耐药肿瘤模型的抑瘤能力评价中,化合物25在剂量为30 mg · kg-1时便能实现肿瘤缓解,而对照药GDC-0810剂量达100 mg · kg-1时才表现出同等效果[33]。
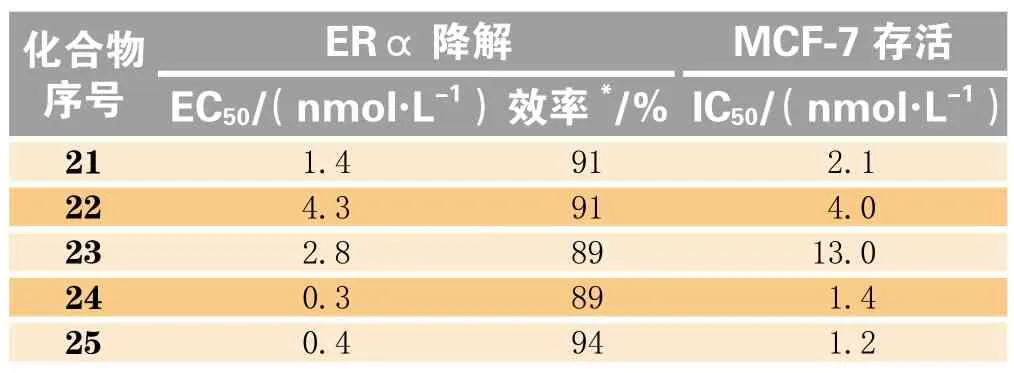
表6 基因泰克公司第2次修饰部分化合物体外数据Table 6 In vitro activity data of some compounds obtained from second-round modification by Genentech
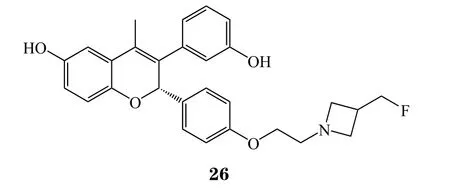
化合物GDC-0810的Ⅱ期临床试验于2017年4月被罗氏终止。虽然具体终止原因并未披露,但是已知GDC-0810在大鼠子宫内膜细胞中具有微弱的激动剂作用,与他莫昔芬相似,其具有提高患者子宫内膜癌患病风险的隐患。对于基因泰克及其母公司罗氏来说,GDC-0810可能已经不具备在SERDs领域竞争的优势,故其临床试验被终止。但是基因泰克并未放弃SERDs药物的开发。在第255届美国化学学会全国会议暨博览会上,其披露了在SERDs药物开发项目上继续推进的分子GDC-0927(26)的具体结构。目前,该化合物正处于Ⅰ期临床试验中(试验代号:NCT02316509)。
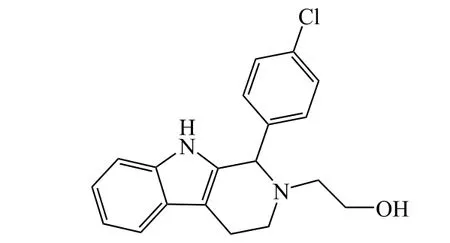
3.4 艾拉司群
起初,卫材药业在研发一款SERMs药物,用于治疗潮热、盗汗等更年期妇女经历的血管舒缩症状。卫材得到一系列化合物(见图8通式[34])。当Y为氮原子时,该系列化合物都具有较好的ER结合能力。其中艾拉司群(elacestrant,RAD-1901,27)因其在小鼠潮热模型中表现出较好的降低体温的能力,卫材将其作为重点研发对象。根据其作用机制,当时的艾拉司群只能被定义为SERMs。然而在对艾拉司群的深入研究中,方圆健康(Radius Health)公司发现了艾拉司群具有下调ER水平的能力。此后,艾拉司群被正式归类为SERDs。方圆健康也开展了一系列实验,评估艾拉司群作为SERDs下调ER水平以及抑制癌细胞增殖能力。实验结果显示:艾拉司群在MCF-7细胞系中降解ERα EC50为 0.6 nmol · L-1,抑制 MCF-7 细胞增殖 IC5为 4.2 nmol · L-1,与 AZD9496、GDC-0810 等均处于同一水平[35]。
在体内药效评估中,60 mg · kg-1艾拉司群在给药后第40 d肿瘤生长抑制率达88%,与对照组氟维司群(0.5 mg · d-1)的抑瘤效果相当;更高剂量艾拉司群抑瘤作用的实验结果显示,90和120 mg · kg-1艾拉司群给药后42 d,肿瘤生长抑制率达96%以上,明显优于氟维司群且耐受良好[35]。
Garner等[35]认为,高剂量艾拉司群在小鼠体内达到的血药浓度能在人体中实现。合理推测,当艾拉司群应用于临床时产生的抑瘤能力会强于氟维司群现有的给药方案。
艾拉司群已于2017年10月获得FDA批准的快速审查资格。目前其正处于治疗ER阳性、Her-2阴性晚期或转移性乳腺癌的Ⅰ期临床试验。其临床试验(NCT02650817)反馈的FES-PET数据显示,73%的实验对象ER阻断程度大于75%,优于氟维司群的临床表现
此外,方圆健康还开展了SERDs与其他抗肿瘤药物联用的研究,包括艾拉司群与mTOR抑制剂依维莫司以及CDK4/6抑制剂帕博西尼的联合用药[36],结果显示联合用药相对于艾拉司群单药具有更好的抑瘤效果。
2016年1月,方圆健康宣布与诺华展开合作,评估艾拉司群与CDK4/6抑制剂瑞波西利(ribociclib)联合使用的安全性以及临床疗效。目前该治疗方案正处于Ⅰ期临床试验中。
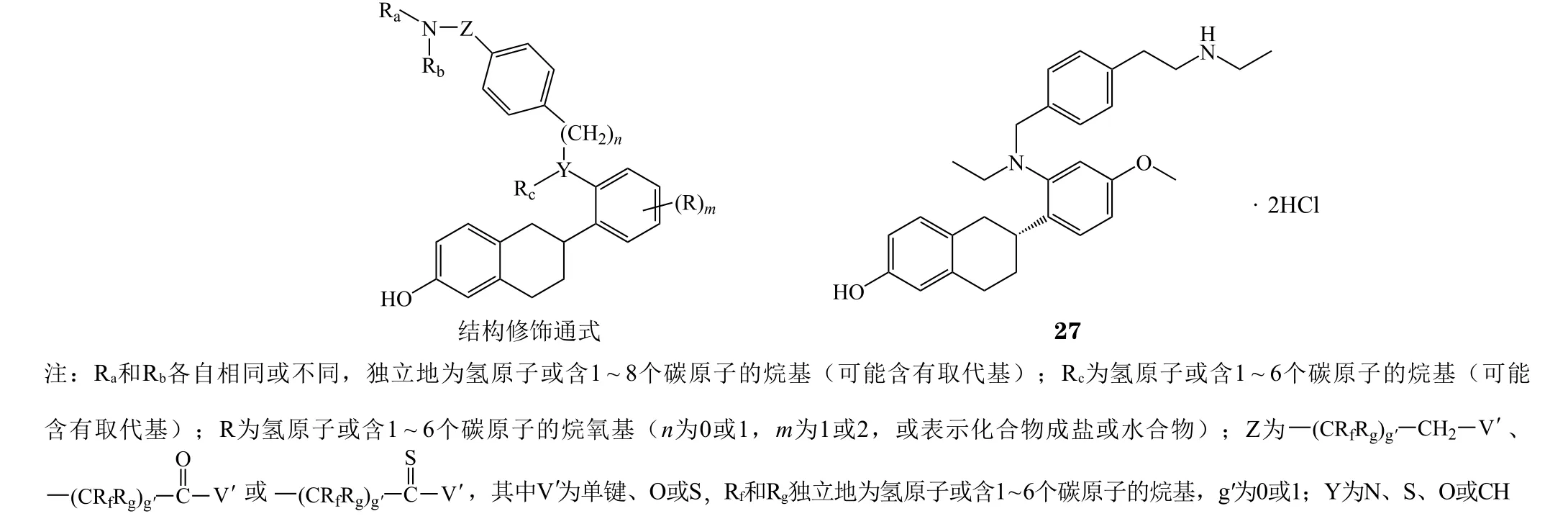
图8 卫材药业化合物结构修饰通式及化合物27Figure 8 The general modification structure patented by Elisa and the structure of compound 27
4 结语
以ER为靶点的乳腺癌内分泌治疗药物根据作用机制不同分为SERMs、SERDs以及SERCAs等3种。虽然SERMs类药物在临床上的使用已有数十年的历史,但尚未开发出治疗乳腺癌的理想SERMs药物。即使改善了他莫昔芬的部分缺陷,第2、3代SERMs药物仍旧受到疗效欠佳以及交叉耐药等因素困扰,无法得到更多的临床应用。SERCAs药物的研发则正处于萌芽阶段,其共价结合的作用方式也可能在药物毒性方面产生重要影响。而对于SERDs药物来说,虽然已经上市的仅有氟维司群,但其在抑制乳腺癌癌细胞增殖能力上已得到了临床试验的验证,优于他莫昔芬;并且其下调ER水平的机制也不会与他莫昔芬产生交叉耐药,对于他莫昔芬耐药的乳腺癌患者同样有效。当然,氟维司群的缺点同样明显。因此各大制药公司紧盯以ER为靶点的内分泌治疗药物市场,纷纷加入SERDs药物的研发,改善氟维司群缺点,以期得到一个口服生物利用度较好的SERDs药物。目前,以AZD9496为代表的口服SERDs药物大部分已进入临床试验阶段。这些化合物通过改善口服生物利用度,提高血药浓度,在临床前都表现出优于氟维司群的ER降解以及抑瘤能力,Ⅰ期临床也未有相关重大不良反应的报道。阿斯利康在拥有唯一已上市SERDs药物的基础上,在SERDs药物的开发处于领跑状态。未来几年,开发口服生物利用度高、临床疗效更好的SERDs药物,将成为乳腺癌内分泌治疗领域的研究热点。
猜你喜欢
物流技术与应用(2022年8期)2022-08-26
基层中医药(2020年5期)2020-09-11
物流技术与应用(2020年4期)2020-05-06
小小艺术家(2019年8期)2019-11-21
天津医科大学学报(2019年6期)2019-08-13
实用肿瘤学杂志(2018年3期)2018-01-31
中国卫生标准管理(2014年2期)2014-01-31
中国合理用药探索(2012年2期)2012-03-20
中国合理用药探索(2011年9期)2011-03-20
中国合理用药探索(2011年7期)2011-03-20
