
查尔酮衍生物在阿尔茨海默病治疗和诊断中的研究进展
2019-06-06 07:40:40陈琳王柯人桑志培
药学进展 2019年4期
陈琳,王柯人,桑志培*
(1.杭州市余杭区第一人民医院药剂科,浙江 杭州 311100;2. 南阳师范学院化学与制药工程学院,河南 南阳 473000)
阿尔茨海默病(Alzheimer's disease,AD),又称早老性痴呆,是老年人群中致死率和发病率最高的疾病之一。目前全球有超过4 700万痴呆症患者,预计到2050年将达到惊人的1.315亿,其中中国痴呆症患者将达到3 000万[1-2]。截至目前,美国FDA批准用于AD的临床治疗药物主要包括胆碱酯酶抑制剂(卡巴拉汀、他克林、多奈哌齐和加兰他敏)和N-甲基-D-天门冬氨酸(N-methyl-D-aspartic acid,NMDA)受体拮抗剂美金刚,长期临床应用表明,上述药物仅能在短时期内局限性地改善患者症状,并不能有效阻止或逆转病程[3]。AD病因复杂,涉及的关键靶点有25个[4]。目前单靶点药物不能从根本上改变AD的进程,针对诱发AD的多个靶点、多个环节同时干预疾病的多靶点药物,即“一药多靶(one-drug-multiple-targets)”有望为AD治疗带来新的曙光[5]。
β淀粉样蛋白(amyloid β-protein,Aβ)级联假说揭示了Aβ沉积是导致AD的重要原因,在病理状态下Aβ通过高度β折叠形成不溶性纤维并产生沉积,从而发挥神经毒性,进一步引起炎症反应和氧化应激,并导致神经递质的缺乏和认知障碍,最终导致AD并发。另外,研究表明:Aβ斑块在脑内的缓慢沉积是AD的重要病理特征之一,而尸检在脑部发现Aβ斑块也是AD确证的重要标准。因此,以Aβ斑块作为靶点,开发与之具有高亲和力的分子探针,并利用分子影像技术实现AD的早期无损伤诊断具有重要的临床和现实意义[6]。
查尔酮的化学结构为1,3-二苯基-2-丙烯-1-酮(1)以其为母体的天然化合物多存在于甘草、红花等植物中,是植物体内合成黄酮和异黄酮的主要前体物质[7]。由于其分子结构具有较大柔性,能与多种生物大分子相互作用,因而呈现出广泛的生物活性[8]。本文对查尔酮衍生物在AD治疗和诊断方面的研究进展作一综述。
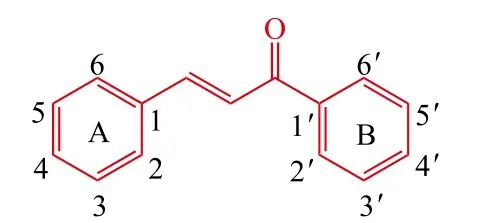
1
1 阿尔茨海默病治疗药物
查尔酮具有多重药理活性,其中抗炎、抗氧化、清除自由基和抑制Aβ聚集等活性对AD具有潜在的治疗作用,科研工作者基于经典药物设计原理和多靶点药物设计策略对查尔酮母体结构进行结构优化,发现了一系列抗AD先导化合物。
1.1 乙酰胆碱酯酶抑制剂
Sheng等[9]设计并合成了一系列查尔酮衍生物作为潜在的乙酰胆碱酯酶(acetycholinesterase,AChE)抑制剂,测试结果显示合成的查尔酮衍生物均为选择性AChE抑制剂。构效关系表明:取代基位于A环的4位比3位显示了更好的抑制活性;取代基对AChE的抑制活性也有一定的影响,其活性顺序由大到小依次为吡咯、二乙胺、哌啶、N-乙基-甲基胺,其中化合物2是活性最强的高选择性AChE抑制剂(IC50= 0.037 μmol ·L-1),为进一步开发抗AD药物提供了重要线索。
2
Liu等[10]设计合成了一系列新颖的胺烷基取代的F-查尔酮衍生物,F原子的位置及胺烷基显著地影响了化合物对胆碱酯酶的抑制活性。构效关系研究表明:3-氟取代的查尔酮衍生物的抑制活性比2-氟取代或4-氟取代显示了较低的AChE抑制活性,当胺烷基为二乙胺或二甲胺时,2-氟取代查尔酮显示了最强的AChE抑制活性,当胺烷基为哌啶或吡咯时,4-氟取代查尔酮显示了最强的抑制活性;另外,氟原子的位置对于胆碱酯酶的选择性具有重要影响,4-氟取代的查尔酮衍生物对于AChE抑制具有更高的选择性,其中化合物3显示了最强的 AChE 抑制活性 [IC50= 0.21 μmol · L-1,选择性指数(selectivity index,SI)为65]。酶动力学和分子对接研究表明:化合物3是一个混合型AChE抑制剂,能够同时作用于AChE的催化活性部位(catalysis active site,CAS)和外周阴离子位点(peripheral anionic site,PAS),其可作为抗AD药物研发的候选化合物。
3
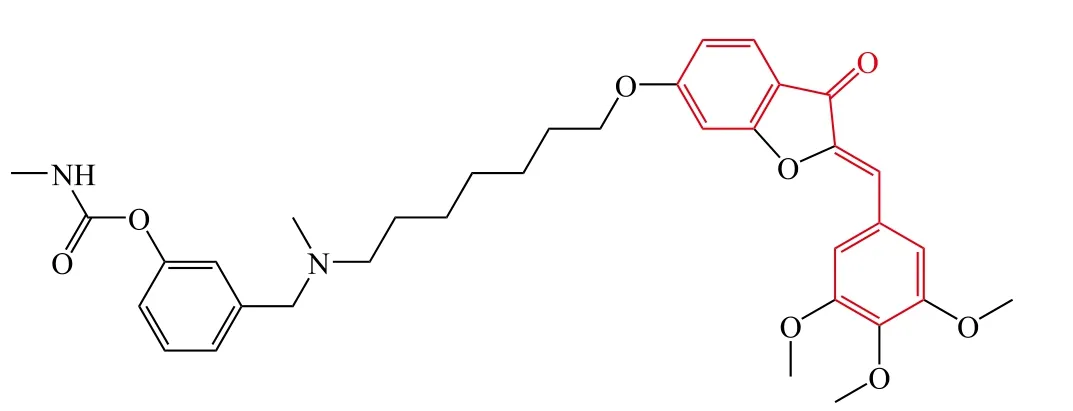
Belluti等[11]合成了一系列噢哢类查尔酮-O-氨基甲酸酯衍生物,研究结果表明含有7个亚甲基的化合物绝大部分显示了纳摩尔水平的AChE抑制活性,其中拥有3,4,5-三甲氧基苯基结构的化合物4的AChE抑制活性最强(IC50= 0.52 nmol · L-1),且对丁酰胆碱酯酶(butyrylcholine esterase,BuChE)也有良好的抑制活性(IC50= 136 nmol · L-1)。构效关系研究表明:当用α-或β-萘基、蒽基、苯基取代化合物4中的3,4,5-三甲氧基苯基时,AChE抑制活性则明显降低,当取代为3,5-二氯苯基片段时,AChE抑制活性进一步减弱;当化合物4中呋喃环被环戊酮、吡喃酮和环己酮取代时,AChE抑制活性不同程度地降低。另外,化合物4能够抑制AChE诱导的Aβ聚集,上述研究结果为寻找新型的抗AD药物提供了重要支撑。
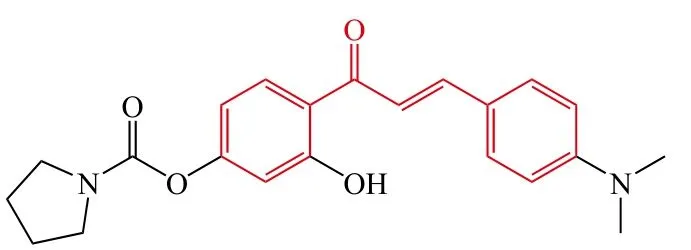
Rampa等[12]在化合物4的基础上,通过呋喃环开环设计并合成了一系列氨基甲酸酯类化合物,生物活性测试结果表明碳链长度对AChE抑制活性有显著的影响,当碳链长度为3 ~ 7时,目标化合物显示了良好的 AChE 抑制活性,IC50达 0.81 ~ 1.80 nmol·L-1,然而,当碳链长度增加到10时,AChE抑制活性则显著降低。化合物5的AChE抑制活性最强(IC50= 0.81 nmol · L-1),BuChE抑制活性也较强(IC50= 106 nmol · L-1)。另外,化合物5对Aβ诱导的神经毒性还有一定的保护作用,同时能够调节神经炎症反应,为抗AD药物研发提供了先导化合物。
Liu等[13]设计并合成了一系列含氮的查尔酮衍生物,体外生物活性测试的构效关系表明:查尔酮衍生物的胺烷基侧链对AChE抑制活性有显著的影响,其中化合物6的AChE抑制活性最强(IC50= 0.85 μmol · L-1),并且对BuChE具有最高的选择性(SI = 35.79)。与化合物3相似,化合物6也是混合型AChE抑制剂,能够同时作用于AChE的CAS和PAS部位,是AD治疗的潜在的先导化合物。
4
5
6
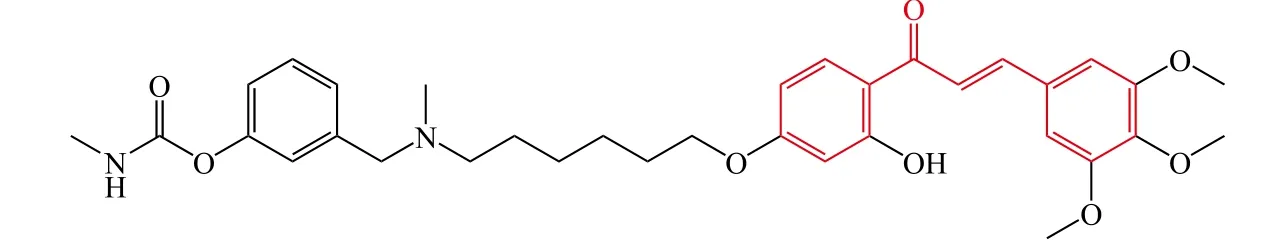
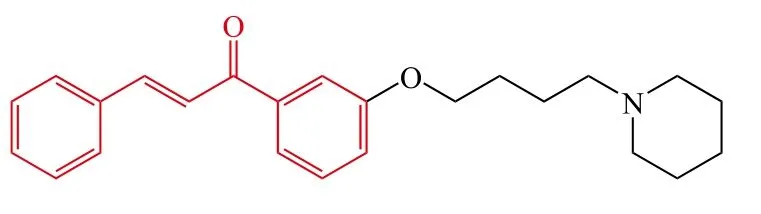
Wang等[14]设计并合成了一系列新颖的查尔酮-卡巴拉汀衍生物。体外生物活性测试结果表明:查尔酮骨架上含有2个氨基甲酸酯(2′和4′位)时,对胆碱酯酶几乎没有抑制活性,当单氨基甲酸酯位于查尔酮3′位时对胆碱酯酶也几乎没有抑制活性,单氨基甲酸酯位于 1′、2′、5′和 6′时则为高选择性的 BuChE 抑制剂。氨基甲酸酯片段中的取代基对胆碱酯酶抑制活性也有一定的影响,一般来说,N-乙基-N-甲基胺和二甲胺取代时显示了较高的胆碱酯酶抑制活性,其中化合物7对AChE和BuChE均有显著的抑制活性,其IC50分别为 0.87 和 0.36 μmol · L-1。另外,化合物 7 能够降低SH-SY5Y细胞中氧化应激的产生。化合物7是一个潜在的先导化合物,其进一步的结构优化仍在进行中。随后,Xiao等[15]对查尔酮-卡巴拉汀衍生物中查尔酮的4′位进行结构改造,得到了一系列新颖的4′-氨基查尔酮-卡巴拉汀衍生物。研究结果表明:该类目标化合物为选择性AChE抑制剂,且查尔酮母核的4′位被环胺取代比非环胺取代显示了更强的抑制活性,其中4′位被吡咯环取代得到的化合物8的AChE抑制活性最强(IC50= 4.91 μmol · L-1),并对 BuChE具有很高的选择性。酶动力学和分子对接结果表明化合物8是一个混合型AChE抑制剂,能够同时结合AChE的CAS和PAS部位。另外,化合物8显示了较强的抗氧化活性[氧化自由基吸收能力(oxygen radical absorbance capacity,ORAC)为水溶性维生素E的2.83倍]和金属离子络合能力,并且能够抑制自身诱导和Cu2+诱导的Aβ1-42聚集,其抑制率分别为89.5%和79.7%,同时化合物8还是一个高选择性的单胺氧化酶(MAO)-B抑制剂(IC50=0.29 μmol · L-1)。因此,化合物8是一个潜在的抗AD先导化合物。
1.2 神经保护剂
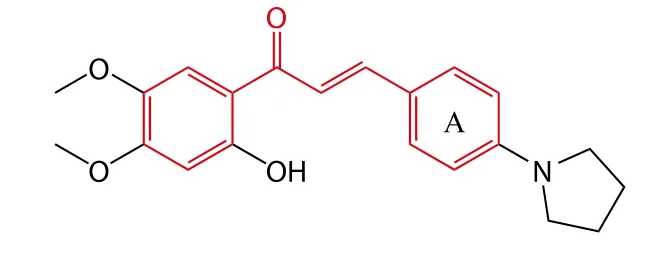
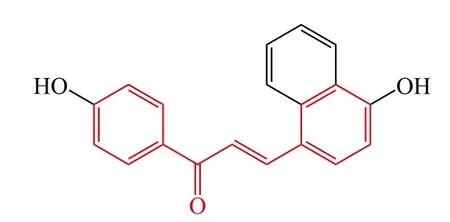
Kim等[16]设计并合成了一系列新颖的查尔酮衍生物,并测试了抗神经退行性变化的保护作用,研究结果表明:化合物9在10 μmol · L-1浓度下显示了最强的自由基清除能力,对Aβ诱导的神经细胞损伤显示了显著的保护作用,对于Aβ1-42注射小鼠,体内实验表明化合物9在20和50 mg · kg-1时能够提高学习和记忆能力对神经退行性疾病是一个潜在的治疗药物。
1.3 单胺氧化酶B抑制剂
MAO是催化单胺类物质氧化脱氨反应的酶,调节大脑和外周组织主要单胺类神经递质(如5-羟色胺、去甲肾上腺素、多巴胺和苯丙胺等)的浓度和新陈代谢其包括MAO-A和MAO-B,MAO-A主要位于神经元轴突,MAO-B主要位于胶质细胞。选择性MAO-A抑制剂在临床上用作抗抑郁和抗焦虑药,抑制MAO-B可改善AD的症状[17-19]。选择性的MAO-B抑制剂(如司来吉兰和雷沙吉兰)已经证实能够延缓AD的神经变性[20]。因此,选择性MAO-B抑制剂也是治疗AD的重要策略。
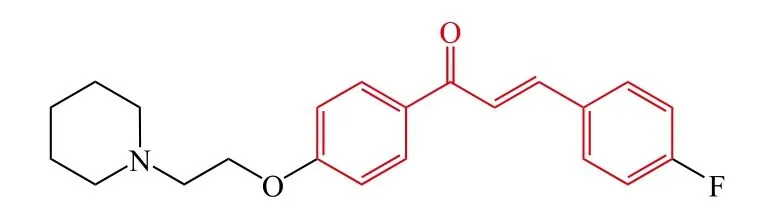
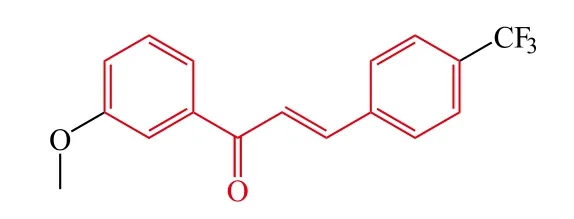
Mathew等[21]合成了一系列甲氧基查尔酮,并测试了其对MAO-A和MAO-B的抑制活性,研究结果表明该系列化合物均是可逆的选择性MAO-B抑制剂,其中化合物10的MAO-B抑制活性最强(IC50=0.22 μmol · L-1,SI = 0.05),分子对接进一步为其高活性提供了合理的解释,化合物10作为可逆的选择性MAO-B抑制剂,为进一步的抗AD药物的结构优化奠定了基础。
Morales-Camilo等[22]合成并测定了查尔酮和噢哢类化合物的MAO抑制活性,研究结果表明化合物11显示了最强的、高选择性的MAO-B抑制活性(IC50= 2.8 μmol · L-1,SI>35.7),其分子对接结果进一步证实了其良好的 MAO-B 抑制活性。
1.4 μ-钙蛋白酶和组织蛋白酶B双靶点抑制剂
μ-钙蛋白酶(μ-calpain)是一种钙依赖的半胱氨酸蛋白酶,能够被体外微摩尔水平的钙离子浓度激活,细胞内钙离子的紊乱导致了μ-钙蛋白酶的高度活化,进一步通过β-分泌酶的高表达增加了Aβ的聚集和Tau蛋白磷酸化[23]。研究表明组织蛋白酶B(cathepsin B)在神经炎症的起始阶段和神经功能紊乱中起着关键作用[24]。因此,抑制μ-钙蛋白酶和组织蛋白酶B是治疗AD潜在的治疗手段。
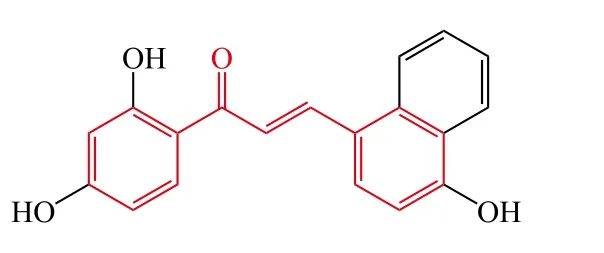
Jeon等[25]设计并合成了一系列查尔酮衍生物,通过测试目标化合物μ-钙蛋白酶和组织蛋白酶B的抑制活性,发现化合物12对两者显示了较强的抑制活性,其 IC50分别为 18.83和 6.34 μmol · L-1。化合物 12在 5 μmol · L-1时能够显著地保护 H2O2诱导的 SH-SY5Y细胞损伤;此外,其还能减少Tau蛋白磷酸化和不可溶Aβ蛋白的形成。综上所述,化合物12是μ-钙蛋白酶和组织蛋白酶 B双靶点抑制剂,对于AD治疗是一个潜在的先导化合物。
1.5 多靶点抑制剂
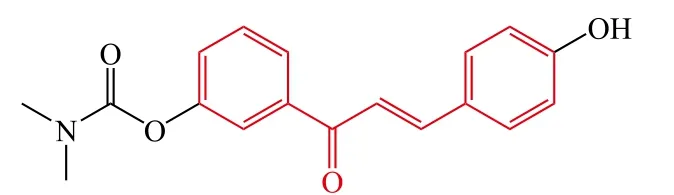
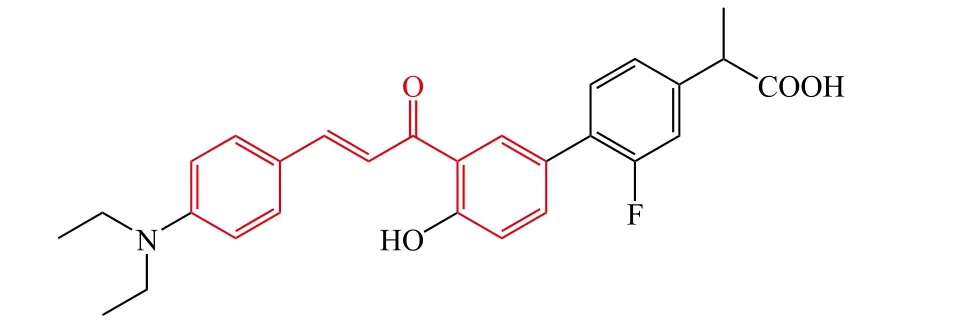
Cao等[26]设计并合成了一系列4′-OH氟比洛芬-查尔酮衍生物作为抗AD潜在的多靶点抑制剂,体外生物活性测试结果表明:大部分目标化合物显示了良好的多重活性,其中化合物13对自身诱导和Cu2+诱导的Aβ1-42聚集显示了最强的抑制活性,其抑制率分别为78.2%和95.0%。此外,其还显示了良好的抗氧化活性、MAO抑制活性、金属离子螯合活性和抗炎活性。更进一步的研究表明该化合物能够透过体外血脑屏障。综上所述,化合物13是一个潜在的具有多靶点性质的抗AD先导化合物,值得进一步开发。
7
8
9
10
12
13
2 β淀粉样蛋白荧光探针
Aβ斑块的沉积是AD发展早期阶段重要的病理特征,针对Aβ的生物标记物——荧光探针将能够有效识别个人是否患AD,对AD早期诊断具有重要的临床意义。
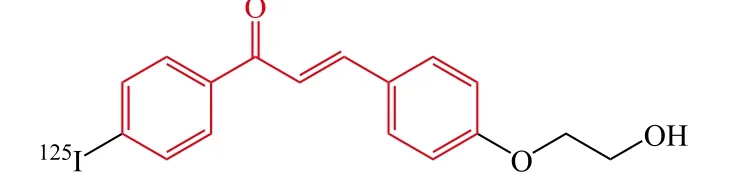
Ono等[27]合成了一系列新颖的查尔酮衍生物,Aβ聚集的体外结合试验表明查尔酮衍生物对Aβ具有良好的结合能力,Ki为 2.9 ~ 104.7 nmol · L-1。通过给正常小鼠体内注射放射性碘化的查尔酮衍生物来研究体内分布情况,结果显示放射性碘化的查尔酮衍生物在脑部有 很 好 的 吸 收 [第 2 min,2.0 ~ 4.7 %ID · g-1(%ID · g-1表示单位质量组织的百分注射剂量)],并且在脑部能够快速地清除(第 30 min,0.2 ~ 0.6 %ID · g-1)。其中化合物14对Aβ1-42聚集显示了最强的结合能力(Ki=2.9 nmol · L-1),其放射性碘化的化合物[125I]14在脑部具有良好的吸收(第2 min,2.04 %ID · g-1),并且在脑部具有良好的清除能力(第2 min,0.61 %ID · g-1)。以上结果提示放射性碘化的查尔酮衍生物对于AD的诊断可能是潜在的荧光探针。
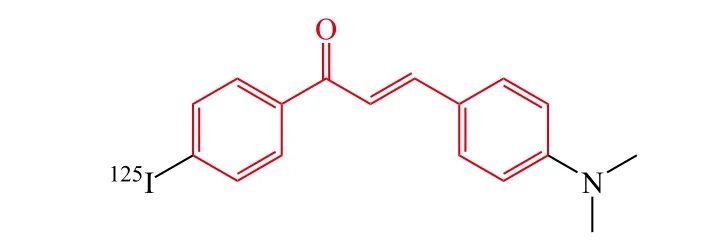
14
同时,Ono等[28]设计并合成了一系列噢哢衍生物作为检测AD患者脑内Aβ斑块的荧光探针。体外Aβ聚集结合实验表明目标化合物对Aβ聚集显示了高亲和力(Ki为 1.2 ~ 6.8 nmol · L-1),其中化合物 15 显示了较好的亲和力(Ki= 6.8 nmol · L-1),体内分布结果表明化合物[125I]15在脑部显示了良好的吸收能力(第2 min,1.89 %ID · g-1),并且能够快速清除(第60 min,0.11%ID · g-1)。因此,噢哢衍生物[125I]15对于AD患者脑内Aβ斑块的检测将是一个很有希望的探针。
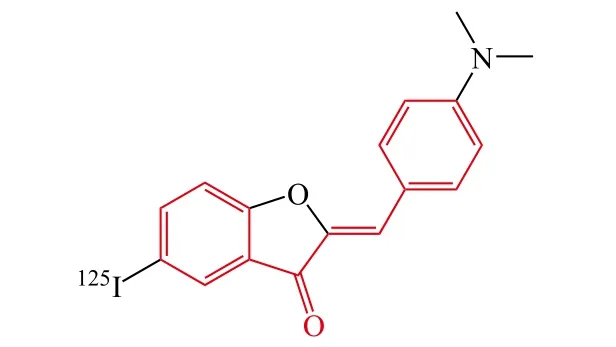
15
随后,Ono等[29]设计并合成了一系列氟代-聚乙二醇(PEG)化的查尔酮用于AD患者Aβ斑块的荧光检测。体外研究结果表明:化合物对Aβ聚集的亲和能力受4位氨基取代基的影响很大,二甲氨基、单甲氨基、氨基对Aβ聚集亲和力依次降低;PEG的长度则对亲和力影响不大,其中含有3个碳链长度的氟修饰的化合物16对Aβ聚集显示了较强的亲和力,其Ki为38.9 nmol ·L-1。小鼠体内生物分布结果表明:[11C]16能够透过血-脑脊液屏障,并且化合物16显示良好的吸收性,其在第2 min的脑部吸收为4.31% ID · g-1,且表现了最快的放射性消除能力(第60 min,0.35 %ID · g-1)。另外,[18F]16在脑部也显示了高的吸收能力和良好的清除能力。整体来说,氟代-PEG查尔酮衍生物16是一个很有希望的Aβ荧光探针。
Ono等[30]设计并合成了4个99mTc标记的查尔酮衍生物作为Aβ荧光探针。研究结果显示该类衍生物对Aβ1-42聚集有更高的亲和力;在正常小鼠体内的分布实验表明:化合物17在脑部显示了最高的吸收能力(第2 min,1.48 %ID · g-1),侧链胺烷氧基部分碳链增加则吸收降低,并且[99mTc]17的放射性在脑部能够快速地清除(第60 min,0.17 %ID · g-1),增加碳链长度则清除能力降低,因此[99mTc]17是一个潜在的Aβ荧光探针
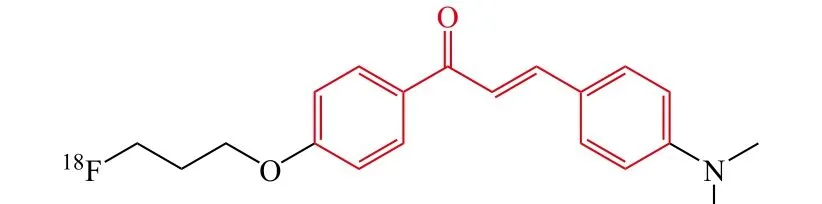
16
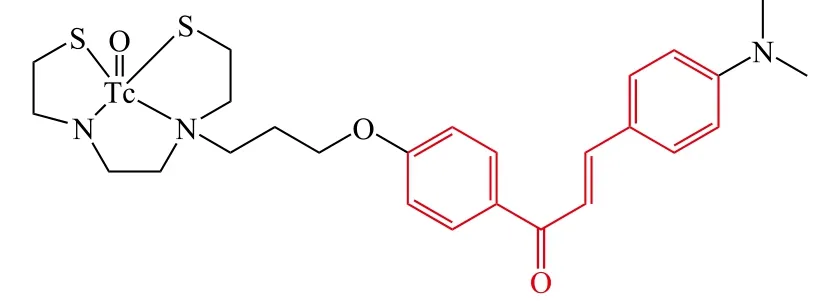
17
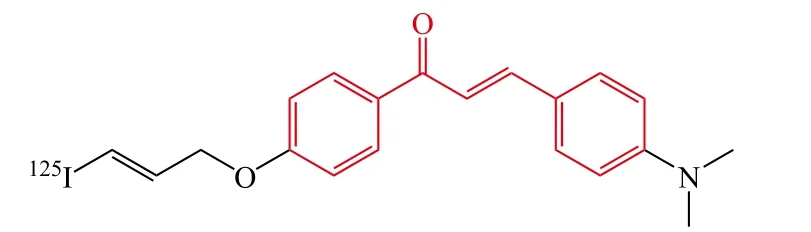
Fuchigami等[31]报道了一系列放射性碘(125I)标记的查尔酮衍生物,用于开发针对Aβ斑块的新型单光子发射计算机断层成像术(single-photon emission computed tomography,SPECT)荧光探针。研究结果表明衍生物18和19对Aβ聚集显示了高亲和力,K分别为24.0和4.5 nmol · L-1。转基因小鼠(Tg2576鼠)脑切片的荧光成像实验表明化合物18和19能够与Aβ斑块特定地结合,体外放射自显影法揭示了[125I]18在Tg2376鼠脑内未观察到Aβ聚集,然而,[125I]19在Tg2576鼠和AD患者脑内观察到了清晰的Aβ斑块聚集。正常鼠的体内分布结果表明:[125I]18表现了更好的药动学性质(第2 min,4.82 %ID · g-1;第60 min,0.45%ID · g-1),然而[125I]19只显示了适当的脑部吸收(第2 min,1.62 %ID · g-1)并能够缓慢地消除(第60 min,0.56%ID · g-1)。整体来说,[125I]19对于Aβ斑块表现了更有前景的结合性质,但需进一步通过结构修饰来提高血-脑脊液屏障透过性以及快速从脑部清除的能力。
18
19
Cui等[32]设计并合成了一系列吲哚-查尔酮衍生物。构效关系表明:苯基对位是卤素(Cl、Br或I)时能够增加化合物对Aβ聚集的结合亲和力;当用羟基取代卤素时则亲和力显著降低,甲氧基取代则使亲和力增强;苯环对位的氨基取代对亲和力也有一定的影响,如二甲氨基取代衍生物和单氨基取代衍生物对Aβ聚集的结合亲和力依次降低;增加芳香共轭体系也能够增加亲和力,其中含有吲哚苯基的查尔酮衍生物20对Aβ聚集表现了较好的亲和能力(Ki= 8.22 nmol · L-1)。进一步的体内生物分布实验表明[125I]20在脑部具有较低的吸收性(第2 min,0.41 %ID · g-1)和缓慢的清除能力(第60 min,0.13 %ID · g-1),该研究为进一步优化吲哚-查尔酮衍生物的结构以便寻找Aβ荧光探针提供了重要线索。
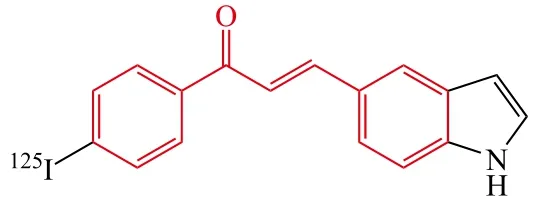
20
3 结语
AD是一种慢性神经退行性疾病,是老年人群中发病率和致死率最高的疾病之一。目前已上市的单一靶点药物只能短期内暂时缓解AD的症状,并不能有效阻止或逆转AD的进程。鉴于AD的复杂病因,多靶点药物(multi-target-directed-ligands,MTDLs)被认为是最有效的治疗策略。Aβ斑块在脑内的缓慢沉积是AD的重要病理特征之一,以Aβ斑块为靶点,开发与之具有高亲和力的分子探针,对AD早期诊断具有重要的临床和现实意义;查尔酮具有重要的药理作用,其衍生物作为多靶点抗AD药物已经取得了新的进展,但由于查尔酮骨架具有α,β-不饱和酮结构,易与体内生物大分子发生结合,使得其作为AD治疗药物受到一定限制。靶向Aβ荧光探针的查尔酮衍生物作为AD早期诊断剂已获得初步的成果,其作为荧光探针的研发将进一步激起科学家的兴趣,值得AD领域深入开发。
猜你喜欢
天然产物研究与开发(2018年9期)2018-10-08 03:25:38
新闻传播(2018年11期)2018-08-29 08:15:30
新闻传播(2018年13期)2018-08-29 01:06:52
中学生数理化·高二版(2016年3期)2016-12-26 09:36:58
新闻传播(2016年9期)2016-09-26 12:20:34
合成化学(2015年10期)2016-01-17 08:56:26
新闻传播(2015年7期)2015-07-18 11:09:57
小猕猴智力画刊(2015年11期)2015-05-30 10:48:04
华东师范大学学报(自然科学版)(2014年1期)2014-04-16 02:54:58
无机化学学报(2014年10期)2014-02-28 17:33:13
