
ABCB11基因突变致暂时性新生儿胆汁淤积症1例病例报告
2019-06-04 07:23田媛媛熊晶晶胡红卫黄永坤
中国循证儿科杂志 2019年2期
田媛媛 刘 梅 熊晶晶 李 檬 曹 佳 胡红卫 黄永坤
1 病例资料
女,2月龄,因“发现皮肤黄染2月”于2018年3月27日入昆明医科大学第一附属医院(我院)儿科。患儿系G2P2,足月剖宫产出生,出生体重3 150 g,生后第3 d发现皮肤黄染,经皮胆红素137~154 μmol·L-1,无发热、激惹、尖叫、抽搐,无少吃、少哭和少动,未予特殊治疗。患儿曾于入我院前1 d因“皮肤黄染未见明显消退”于当地医院就诊,经皮胆红素最高为149 μmol·L-1,总胆红素105 μmol·L-1,直接胆红素94 μmol·L-1,ALT 182.0 IU·L-1,AST 143.0 IU·L-1。病程中,患儿精神、吃奶、睡眠可,尿、粪便的颜色和量均未见异常。
患儿无肝炎病史、服用肝损害药物史和化学毒物接触史。患儿父母体健,否认近亲结婚。母亲孕期诊断为“甲状腺功能减低”,口服左甲状腺素钠片治疗,自述目前甲状腺功能正常。否认家族遗传病史。
入院查体:生命体征平稳,神志清楚,一般情况可。发育正常,营养中等。巩膜、颜面及胸部皮肤黄染,全身皮肤未见抓痕及瘀斑、瘀点,全身浅表淋巴结未触及肿大。双侧瞳孔等大、等圆,对光反射灵敏。咽未见充血、疱疹及脓点。腹平软,按压无哭闹,肝、脾未触及肿大,肠鸣音未见异常。心、肺和神经等系统查体未见阳性体征。
辅助检查:血常规WBC 13.78×109·L-1,淋巴细胞0.75,中性粒细胞0.15,Hb 111.0 g·L-1,PLT 476×109·L-1。血清促甲状腺激素、甲状腺素、三碘甲状原氨酸、游离甲状腺素、游离三碘甲状原氨酸、抗甲状腺球蛋白抗体、抗甲状腺过氧化物酶抗体均未见异常。EB-DNA、CMV-DNA阴性。甲型肝炎抗体测定、乙型肝炎表面抗原、乙型肝炎表面抗体、e抗原、e抗体、核心抗原、核心抗体、核心IgM抗体、表面前S抗原,丙型肝炎抗体,丁型肝炎抗体、戊型肝炎测定均未见异常。血氨136.0 μmol·L-1,乳酸9.40 mmol·L-1。串联质谱显示:CO(游离肉碱,μmol·L-1)70.24 (9.0~50.0)、CO/[十六烷酰基肉碱(C16)+十八烷酰基肉碱(C18),μmol·L-1]54.76 (3.0~45.0)。腹部B超示,肝、胆、胰、脾、双肾和胆管显示均未见异常。磁共振胰胆管成像(MRCP)示,肝左叶稍大,獭尾肝可能;脾稍大;胆囊、胰腺平扫未见异常。
患儿入院排除胆道梗阻后予保肝、熊去氧胆酸利胆等治疗,表1显示,入院第9 d时ALT和AST较前明显下降,但胆红素、总胆汁酸(TBA)未下降或下降不明显,加用多巴胺、山莨菪碱扩张胆管、促进胆汁排泄,入院第15 d时AST、ALP、GGT、TB、DBIL、IDBIL有所下降,ALT降至正常,患儿皮肤黄染有所减轻,病情好转,带药出院。出院后继续口服联苯双酯、复方甘草酸苷片、熊去氧胆酸胶囊。7月龄时电话随访,患儿肝酶、胆汁酸和胆红素等各项指标均已降至正常,身高、体重、智力较同性别、同龄儿童无迟缓,无皮肤黄染、饮食习惯改变等。
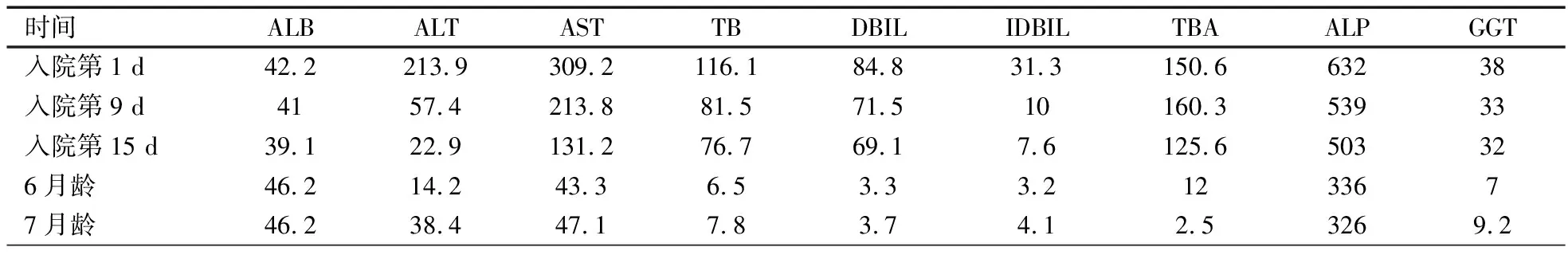
表1 患儿住院随访过程中的实验室检查指标的变化
注 参考值范围:白蛋白(ALB)40~55 g·L-1,ALT 7~40 IU·L-1,AST 13~35 IU·L-1,总胆红素(TB)3.4~17.1 μmol·L-1,直接胆红素(DBIL)<6.8 μmmol·L-1,间接胆红素(IDBIL)1.7~11.1 μmmol·L-1,总胆汁酸(TBA)<10 μmol·L-1,碱性磷酸酶(ALP)45~125 IU·L-1,谷氨酰转肽酶(GGT)7~45 IU·L-1
结合以上情况,考虑患儿为婴儿胆汁淤积性肝病,取得患儿家长同意后予代谢性肝病 panel高通量测序。发现患儿ABCB11基因有2个杂合突变,c.3107C>T(p.S1036F)来自母亲,c.1423T>C(p.C475R)来自父亲,Sanger 测序结果见图1。c.3107C>T和c.1423T>C均为错义突变,在人类基因突变数据库(HGMD)中未见报道,突变位点c.3107C>T经SIFT、PolyPhen_2、 REVEL 预测均为有害。突变位点c.1423T>C经SIFT、PolyPhen_2、 REVEL 预测为无害。
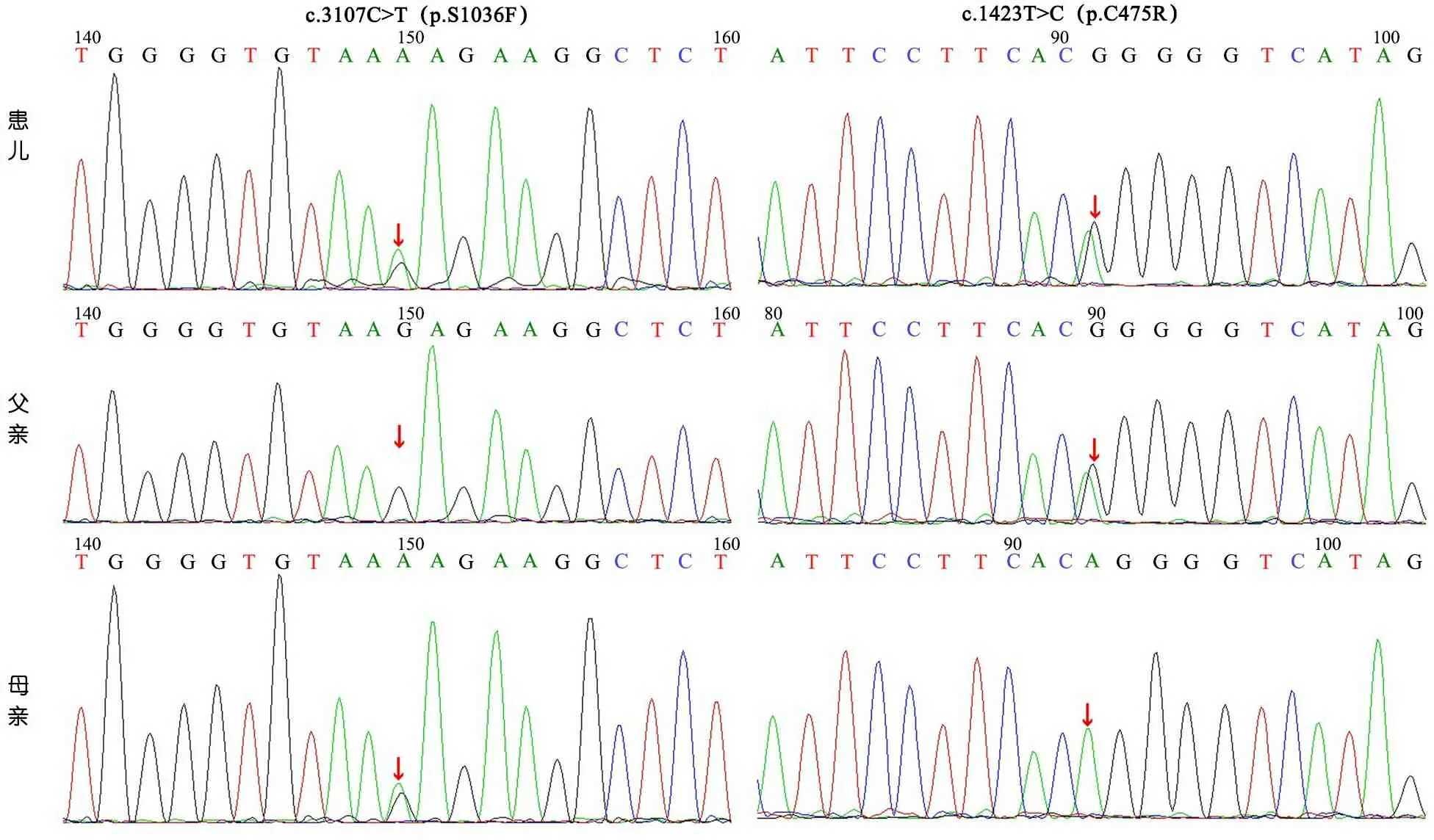
图1 患儿及其父母ABCB11基因Sanger测序结果
注 患儿及其母亲为c.3107C>T(p.S1036F)杂合变异,患儿及其父亲为c.1423T>C(p.C475R)杂合变异
2 讨论
ABCB11基因编码BSEP蛋白。BSEP是ATP结合盒(ABC)转运蛋白超家族B亚族成员之一,几乎均特异性地表达于毛细胆管面的肝细胞膜上,主要功能是与单价胆汁酸盐结合,通过水解ATP将胆盐逆浓度梯度泵入毛细胆管内[1]。ABCB11基因突变可导致进行性家族性肝内胆汁淤积症2型(PFIC2)及良性复发性肝内胆汁淤积症2型(BRIC2)[2],均为常染色体隐性遗传病。PFIC2以严重肝内胆汁淤积为主要特征,多在婴儿早期发病,病情进展迅速,常在儿童或青春期进展为终末期肝病[3]。BRIC2的主要特点为持续数周至数月反复发作的胆汁淤积,一般不会发生进行性肝损伤和肝硬化[4]。本文报告1例ABCB11基因突变携带者,经治疗和随访,其胆汁淤积恢复正常。
本文患儿生后即出现黄疸,且黄疸持续不退,ALT、AST、胆红素和胆汁酸均升高,且直接胆红素/间接胆红素>20%,GGT正常。ABCB11基因存在c.3107C>T(p.S1036F)和 c.1423T>C(p.C475R)杂合错义突变。在HGMD中未见报道,其变异临床意义未明。患儿经保肝及熊去氧胆酸利胆治疗后,7月龄时复查肝功能各项指标均已恢复至正常,患儿胆汁淤积情况未进行性加重,不考虑PFIC2,后期是否会反复发作发展为BRIC2,有待进一步随访,故暂考虑为暂时性新生儿胆汁淤积症(TNC)。
文献复习发现[5-7],ABCB11基因突变已有300多种,涵盖的突变类型有错义突变、无义突变、插入和缺失、剪接位点突变,有报道在欧洲人群中,30%的PFIC2患儿家族检测到p.E297G和p.D482G突变,是欧洲人群中比较常见的2个突变热点,中国人群中未发现热点突变。在已发现的突变位点中,c.1238T>G(Leu413Trp)、c.386G>A(Cys129Tyr)、c.1460G>C(Arg487Pro)、c.725C>T(Thr242Ile)、c.1368delC(del1 bp codon 456)、c.1062T>A(Tyr354Term)、c.2178+1G>C(IVS18 ds G-C +1)等突变位点与PFIC2相关,c.3214-3C>G(IVS24 as C-G -3)、c.1120G>A(Gly374Ser)、c.2594C>T(Ala865Val)、c.2316T>A(Tyr772Term)、c.1211A>G(Asp404Gly)等突变位点与BRIC2相关。已知PFIC2相关突变大多是无义突变或对BSEP功能影响严重的移码突变,而BRIC2相关突变主要是错义突变,对BSEP功能的影响相对较小。
TNC患儿预后良好,约3%的TNC病例可归因于ABCB11基因突变,Liu等[7]对TNC病例的研究发现,c.I416I、c.K436N、c.R928Q和c.IVS7+5G> A等突变位点可能与TNC相关,TNC患儿的ABCB11基因突变谱与PFIC2和BRIC2不同,其对BSEP功能的影响最小。
乳酸为无氧糖酵解的产物,而体内清除乳酸的脏器主要是肝脏,其次为肾脏,肝脏可以通过合成糖原及乳酸经丙酮酸途径进入线粒体氧化供能以清除乳酸,当肝功能障碍时,乳酸清除率下降,至血乳酸水平增高,本例患儿血乳酸水平升高即与肝功能受损有关。
猜你喜欢
今日畜牧兽医(2022年7期)2023-01-05
基层中医药(2022年7期)2022-11-17
现代临床医学(2022年4期)2022-09-29
保健医苑(2022年5期)2022-06-10
珠江水运(2021年15期)2021-08-29
中西医结合肝病杂志(2020年2期)2020-10-27
浙江中医杂志(2019年3期)2019-01-05
祝您健康·文摘版(2018年6期)2018-10-21
黄河黄土黄种人(2017年11期)2017-11-27
水能经济(2017年6期)2017-10-19
