
3β-羟基-Δ5-C27类固醇脱氢酶缺陷1例病例报告
2019-06-04 07:23:22田培超赵彩红史丹丹石小亚
中国循证儿科杂志 2019年2期
张 碧 田培超 赵彩红 王 越 史丹丹 石小亚 姚 运
1 病例资料
女,4岁,因“凝血功能异常伴肝脾肿大3年”于2018-1-8入郑州大学第一附属医院(我院)儿内科。患儿1岁7个月时(2014-9-6)因“头皮血肿反复不愈”至我院就诊,凝血功能指标见表1,肝功能未见异常(表2),考虑“凝血因子K缺乏”,予“肌注维生素K1、输注新鲜冰冻血浆”后头皮血肿消失,凝血功能改善(表1,2014-9-11)。1年前无诱因鼻衄至当地医院就诊,查PLT 65×109·L-1,骨髓细胞学提示PLT减少,肝肾功能指标均在正常值范围,遂至我院。
注 正常参考值:凝血酶原时间(PT) 9.6~14 s,PT% 70%~130%,活化部分凝血活酶时间(APTT)23~35 s, 纤维蛋白原(FIB)2~4 g·L-1, 凝血酶时间(TT)14~21 s, 纤维蛋白原降解产物(FDP)0~5 μg·mL-1,D-二聚体 0~0.55 mg·L-1
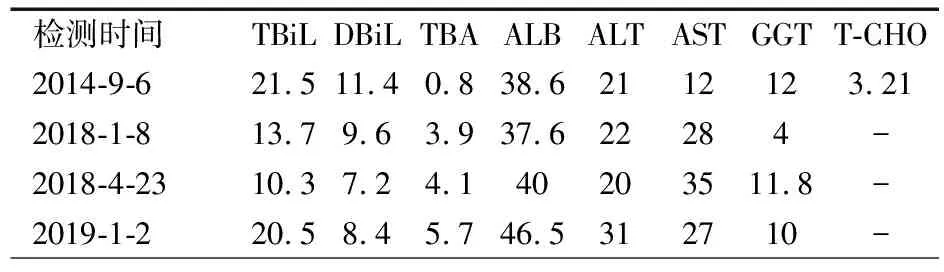
表2 患儿血生化指标检测结果
注 正常参考值:总胆红素(TBiL)0~25 μmol·L-1,直接胆红素(DBiL)0~10 μmol·L-1,总胆汁酸(TBA)0~20 μmol·L-1,白蛋白(ALB)35~55 g·L-1,ALT、AST:0~40 U·L-1,谷氨酰转肽酶(GGT)0~58 U·L-1,总胆固醇(T~CHO)<5.2 mmol·L-1;-:未检测
患儿自起病以来,饮食、睡眠、精神可,体重同正常同龄儿。患儿系G1P1,足月顺产,孕期未诉特殊事件,无窒息抢救史,出生体重2 800 g;生后混合喂养,按时预防接种。父母非近亲结婚,家族中未见同类病患者。
体格检查:体重21 kg,身高110 cm。神志清,精神可,营养、发育良好,皮肤黏膜无黄染、皮疹及出血点。心肺检查未见异常,全腹平软,无肝掌、蜘蛛痣及腹壁静脉曲张,肝脏右锁骨中线肋下扪及4 cm,剑突下扪及4 cm,质地韧,边钝,无触痛。脾脏于左锁骨中线肋下扪及6 cm,质软,未触及异常包块。神经系统查体未见异常。
辅助检查:血常规WBC 6.13×109·L-1,Hb 113 g·L-1,PLT 86×109·L-1。凝血酶原时间(PT)22.2 s,PT% 37.0%,活化部分凝血活酶时间(APTT)50.7 s;肝功能(表2)、肾功能、血糖、血脂均正常;腹部彩超示肝脏肋下4 cm,包膜尚光滑,实质回声增粗;胆壁增厚,内透声差;脾脏厚径35 mm,长径130 mm,肋下65 mm, 实质回声均匀,脾静脉增宽;双肾盏多发强回声。骨髓细胞检查:骨髓增生明显活跃,粒系和红系比值正常,细胞形态大致正常,色素充盈可;全片可见巨核细胞500个,分类25个,幼稚巨核细胞3个,成熟无PLT形成巨核细胞22个,可见成堆及散在PLT。尿常规、尿有机酸气相色谱-质谱联合检测、血氨基酸和酰基串联质谱检测均未见异常胆汁酸代谢产物。
为进一步明确诊断,经家属知情同意后用EDTA抗凝管采集患儿及其父母外周静脉血各2 mL,用DNA提取试盒(北京百泰克生物技术有限公司)提取基因组DNA。经二代测序得到的变异序列,通过8WA软件(版本:0.7.9a)与UCSC hg19参考基因组进行比对,得到致病的突变位点。采用Sanger法验证突变位点及其父母相对应的基因区域,确定患儿的突变类型和遗传方式。图1显示,患儿检出HSD3B7基因c.45-46delAG和c.543dupG的复合杂合变异,分别来自父亲和母亲,符合常染色体隐性遗传规律。前者为编码DNA序列上第45~46位置缺失了A和G 2个碱基,导致p.Thr15fs移码突变;后者为编码DNA第543位上重复插入1个G碱基,导致p.Leu182fs移码突变。在人类基因突变数据库(HGMD)及千人基因组数据库检索,c.543dupG目前尚无报道,经MutationTaster在线分析软件预测该突变为致病性突变;c.45-46delAG已有报道[1]。
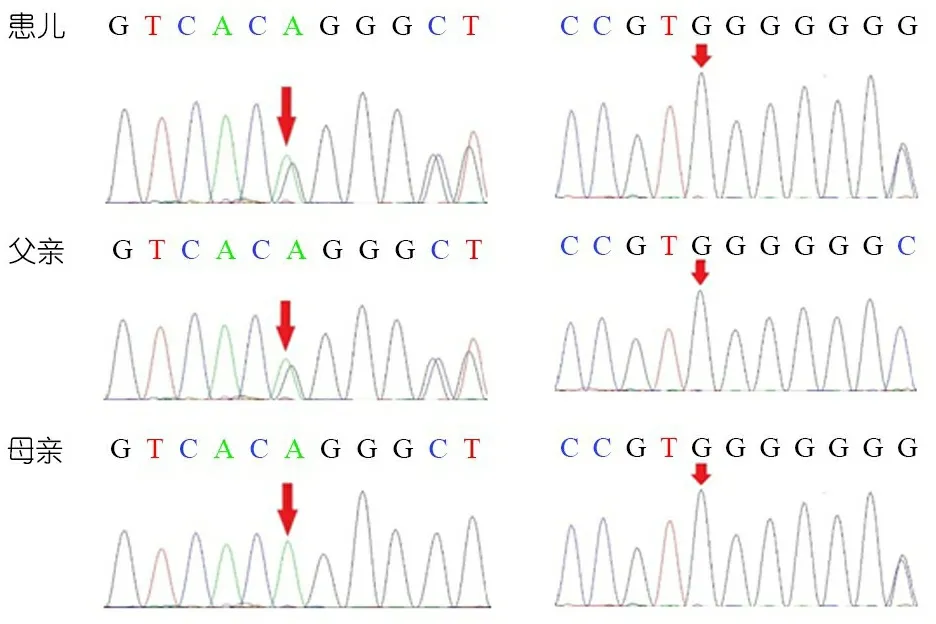
图1 患儿及其父母HSD3B7基因测序结果
注 患儿为c.45-46delAG和c.543dupG的复合杂合变异,患儿父亲携带HSD3B7基因的杂合变异c.45-46delAG,患儿母亲携带1个HSD3B7基因的杂合变异c.543dupG
患儿以PLT减少起病,主要考虑血液系统疾病,未给予特殊治疗。基因确诊3β-羟基-Δ5-C27类固醇脱氢酶缺陷,予鹅去氧胆酸(CDCA)10 mg·kg-1·d-1,tid;脂溶性维生素A、D、E和K。治疗1个月后(表1,2018-4-23)复查凝血功能基本正常。出院后随访至2019-1-2,血常规及凝血功能正常(表1);腹部彩超:肝实质光点增粗,肋下约3 cm,形态正常,脾脏肋下5 cm,双肾皮质强回声。
2 讨论
原发性胆汁酸合成障碍是一种罕见的常染色体隐性遗传性疾病,由于胆汁酸固醇环的修饰和侧链的氧化不全所致,占新生儿及儿童不明原因胆汁淤积症的1%~2%[2, 3]。胆汁酸是在肝脏中由胆固醇合成,涉及17种肝酶[4],其中3β-羟基-Δ5-C27类固醇脱氢酶缺陷是初级胆汁酸合成障碍中最常见的酶缺陷。3β-羟基-Δ5-C27类固醇脱氢酶由位于16号染色体16p11.2的HSD3B7基因编码,是肝脏内胆汁酸合成过程的关键酶之一。
1987年Clayton等[5]首次报道了3β-羟基-Δ5-C27类固醇脱氢酶缺陷。检索万方数据知识服务平台、维普期刊资源整合平台、中国学术期刊网络出版总库和PubMed,国内外报道的3β-羟基-Δ5-C27类固醇脱氢酶缺陷患儿共72例,国内文献14例[1, 6-8],国外文献58例[5, 9-18]。男42例,女30例,平均年龄4.0岁(1月至17岁)。临床表现:黄疸46例,佝偻病 18例,脂肪泻 13例,凝血功能障碍5例;腹部超声:肝大46例 (其中合并脾大10例),肾脏结构异常14例(2例肾结石、6例多囊肾、2例肾脏结构不清、2例肾脏呈囊性变、2例双肾多发囊性变并钙质沉积);在脂肪泻患儿中有7例患儿出现腱反射消失[19]。16例患儿有家族史(其中4例兄妹)。
72例3β-羟基-Δ5-C27类固醇脱氢酶缺陷患儿起病的严重程度不一,Setchell等[4]证实外源性初级胆汁酸会抑制内源性胆汁酸合成途径,从而抑制内源性毒性胆汁酸产生。实际上,HSD3B7基因敲除小鼠有脂肪和脂溶性维生素吸收障碍且无肝病[20],与本文报道的病例类似,但具体机制尚不清楚。脂溶性维生素吸收障碍作为本病的临床特点之一,在临床中常不被重视。本例患儿表现为脂溶性维生素K缺乏引起的凝血功能障碍。3β-羟基-Δ5-C27类固醇脱氢酶缺陷患儿有神经系统累及,7例患儿出现腱反射消失,且均合并脂肪泻,考虑与维生素E缺乏有关,具体机制不详[19]。当体内维生素D缺乏明显时,部分患儿以行走不稳、佝偻病起病[13, 21]。目前尚无维生素A缺乏相关症状报道。
14例肾脏结构异常形成机制尚无明确解释,予初级胆汁酸替代治疗后双肾结构恢复正常[1, 18],考虑是原发病导致的。也有文献报道就诊时年龄大的患儿,更易出现肾脏结构改变,可能与胆汁酸合成障碍时产生的不饱和胆汁酸的慢性毒性有关[1]。本文病例肾脏彩超显示双肾盏多发强回声,以后的随访中会继续关注肾脏的变化,若随着患儿凝血功能及肝脾的好转,肾脏改变也逐渐减轻甚至恢复正常,则说明肾脏结构改变系原发病导致。
目前针对3β-羟基-Δ5-C27类固醇脱氢酶缺陷的治疗大多是经验性的。仅使用CDCA[1, 13]、CDCA+胆酸片(CA)[13]、熊去氧胆酸(UDCA)+CDCA[14-16]、单独使用CA[12, 15],均被认为有效。Gonzales等对13例该病患者进行了长达20余年的随访,发现口服CA是安全有效的长期治疗方法[12, 14]。本文病例予CDCA 10 mg·kg-1·d-1,tid及脂溶性维生素A、D、E、K,1月后复查凝血功能基本正常,1年后复查腹部彩超发现肝脾略有缩小,肾脏未见明显改变。
UDCA作为亲水性胆汁酸,可以降低胆汁酸诱导的肝细胞膜损伤的风险,临床上常首先以UDCA作为治疗胆汁淤积症的经验性用药,且发现临床症状和生化指标改善显著。推测UDCA可能由于利胆作用暂时缓解部分患儿的临床症状,但由于UDCA不能抑制胆固醇7α-羟化酶活性,不能减少异常的胆汁酸生成,不建议长期应用其治疗3β-羟基-Δ5-C27类固醇脱氢酶缺陷。初级胆汁酸(CA及CDCA)作为替代治疗的原发性胆汁酸,可以进入肝肠循环,并通过负反馈抑制胆固醇7α-羟化酶活性,激活负反馈调节胆汁酸合成,抑制肝毒性代谢产物的产生,提供足够大的胆汁酸池刺激胆汁流动,最后增加管腔内浓度,使脂肪和脂溶性维生素正常化吸收。初级胆汁酸替代疗法最重要的作用是恢复对胆汁酸合成的正常生理反馈。
3β-羟基-Δ5-C27类固醇脱氢酶缺陷作为可以被治疗的罕见的遗传性疾病,临床表现较为多样,在婴儿期常以黄疸为首发症状,同时血清中GGT及总胆汁酸正常或减低,伴有肝脾肿大、脂肪泻、生长发育迟缓、佝偻病、凝血功能障碍、肾脏结构改变等,如果不能及早诊断,可能会导致进展性慢性肝病或肝功能衰竭,甚至死亡。在肝硬化及肝衰竭前诊断此病,给予口服初级胆汁酸替代治疗,可使临床症状显著改善甚至临床治愈。分子遗传学检测是目前诊断该病的金标准。在临床工作中若高度怀疑此病,应尽早行基因检测以明确诊断。进一步探索HSD3B7基因突变的基因型与表型之间的关系,可对指导个体化治疗、预后的判定和遗传咨询具有重要意义。
猜你喜欢
广东医科大学学报(2020年6期)2020-02-06 06:00:58
心肺血管病杂志(2018年11期)2018-12-18 01:51:40
猪业科学(2018年8期)2018-09-28 01:27:22
中成药(2018年7期)2018-08-04 06:04:22
中华老年多器官疾病杂志(2016年8期)2016-05-14 07:16:42
中国医药生物技术(2015年4期)2015-12-26 08:26:36
中学生数理化·中考版(2015年12期)2015-09-10 07:22:44
河南医学研究(2014年3期)2014-02-27 14:51:59
食品科学(2013年23期)2013-03-11 18:30:10
中国现代中药(2012年6期)2012-10-30 01:38:18
