
拷贝数变异致高IgM综合征1例并文献复习
2019-06-04 07:23朱晓娜
中国循证儿科杂志 2019年2期
朱晓娜 夏 宇 杨 军
1 病例资料
男,14月龄,因“反复感染1年余,腹胀伴间断发热3个月”于2018年2月就诊于深圳市儿童医院(我院)风湿免疫科。
患儿生后2 d出现呼吸道感染,4月龄腋窝脓肿,5月龄重症肺炎伴巨细胞病毒(CMV)感染,间断给予IVIG治疗,仍出现多次下呼吸道感染,12月龄马尔尼菲青霉菌败血症,13月龄反复腹胀、轮状病毒腹泻,伴生长发育迟缓及肝脾肿大。曾予呼吸机辅助呼吸,利奈唑胺、万古霉素、美罗培南、头孢哌酮舒巴坦、更昔洛韦、伊曲康唑和伏立康唑等抗感染。
既往史:无乙肝、结核等传染病史及接触史,“青霉素”过敏史,无食物过敏史,既往曾行脓肿切开引流术,无外伤史,无异物吸入史。
个人史:患儿系G3P2,足月顺产,出生体重3 350 g,生后母乳喂养至12月龄,6月龄添加辅食,现普食。2月龄会抬头,4月龄会坐,11月龄会站,13月龄尚不能独走、精细抓物和叫爸爸妈妈。未按时预防接种。
家族史:父母体健,否认家族遗传病史,父母非近亲婚配。
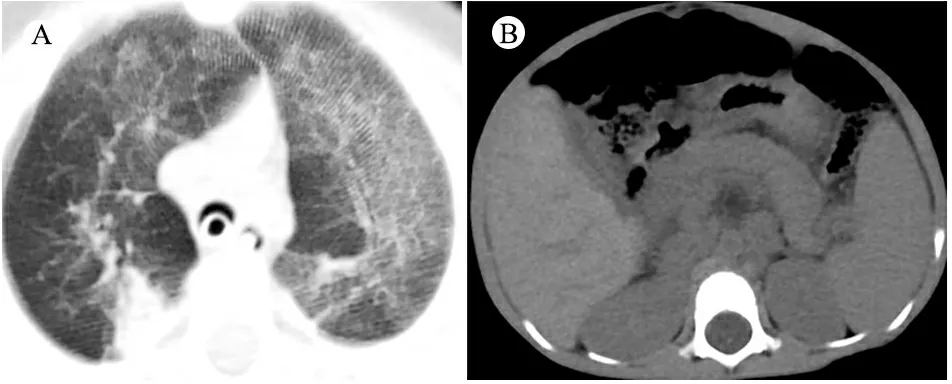
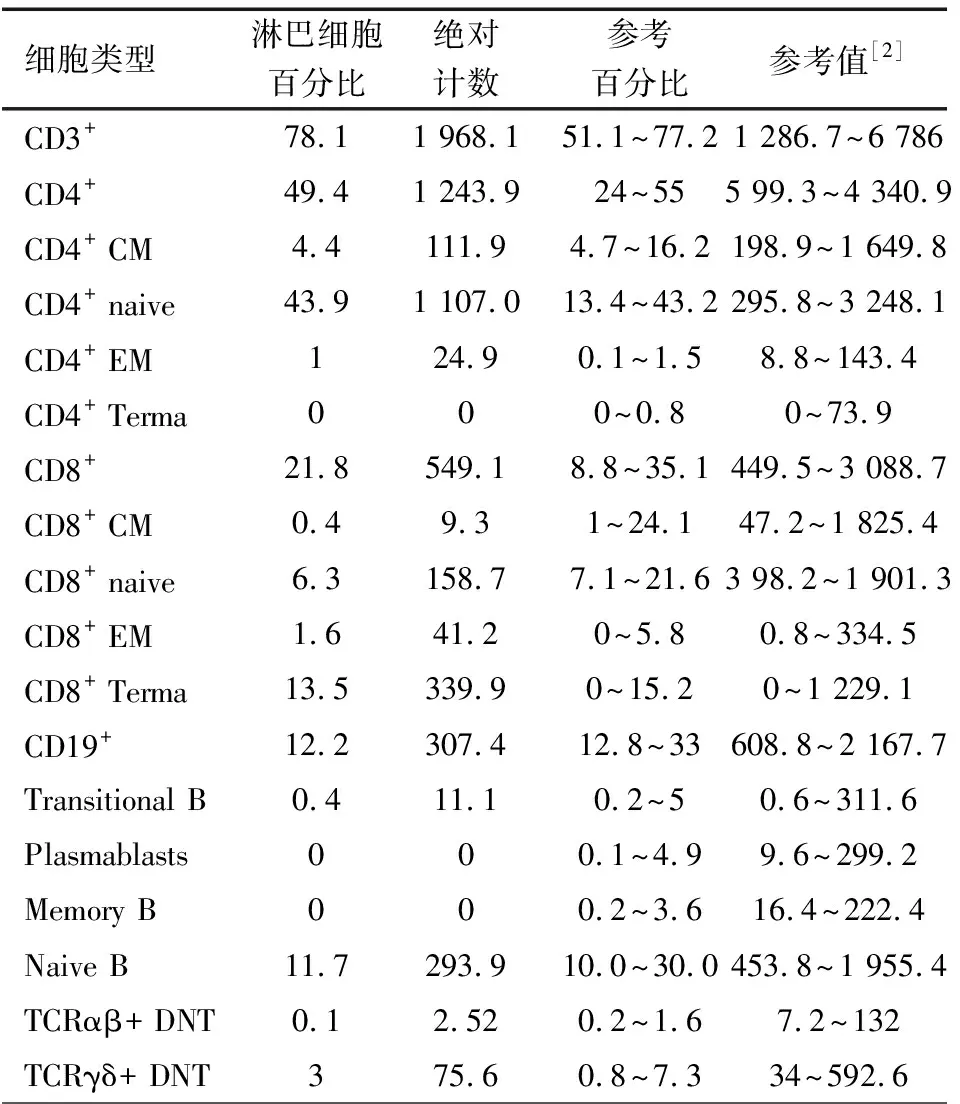
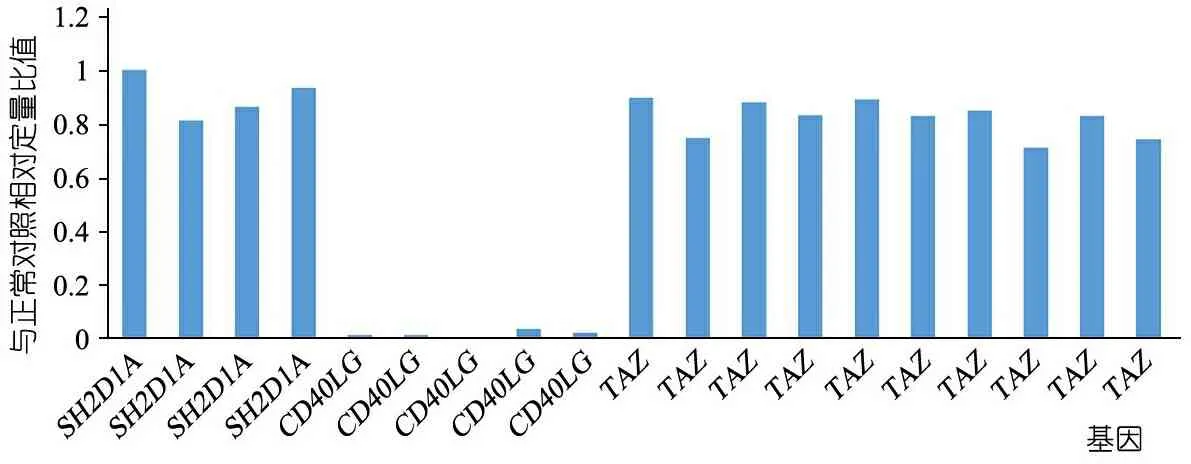
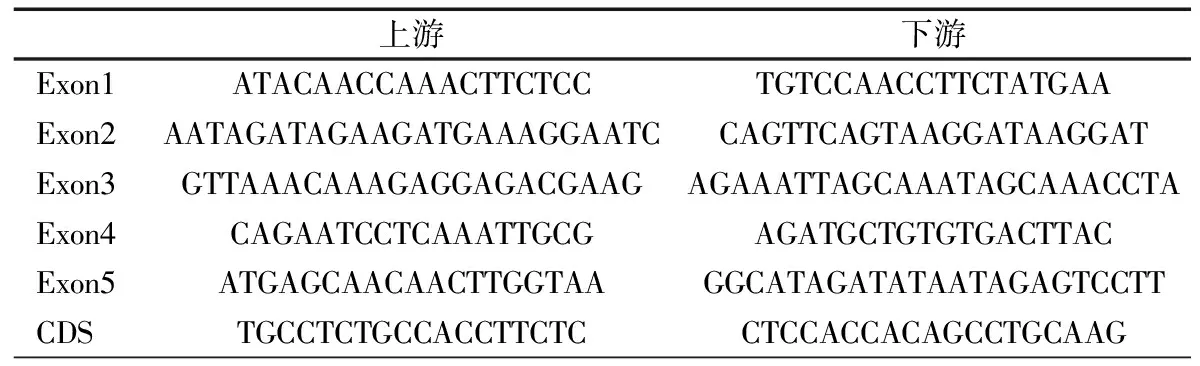
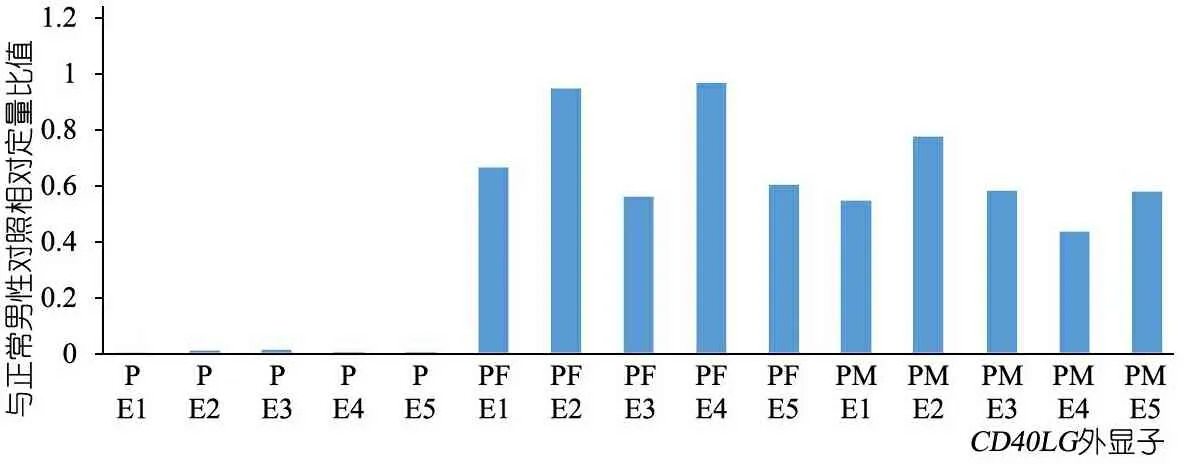
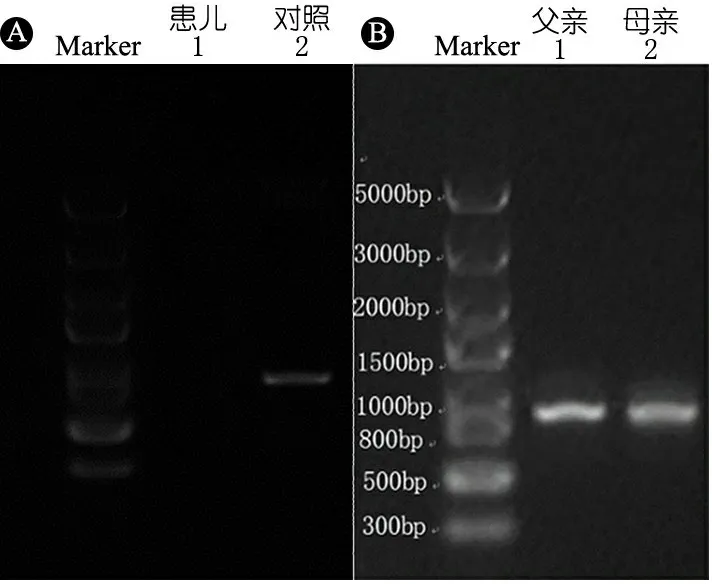
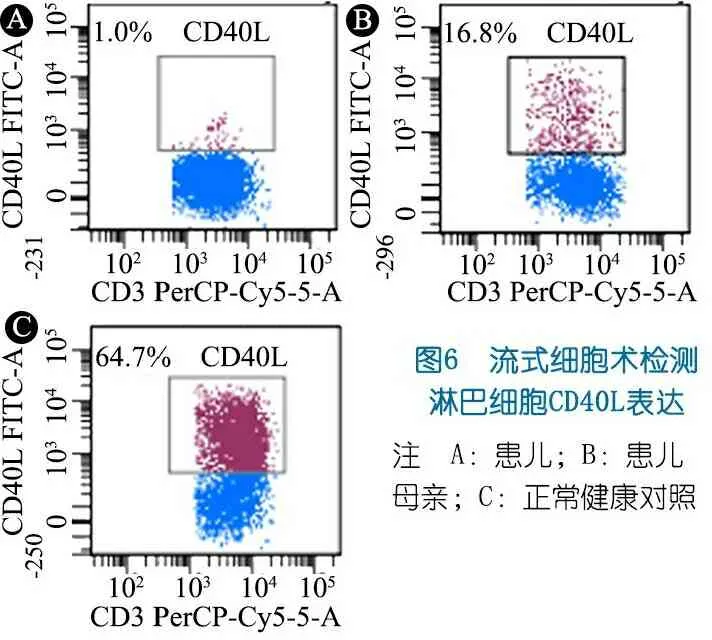
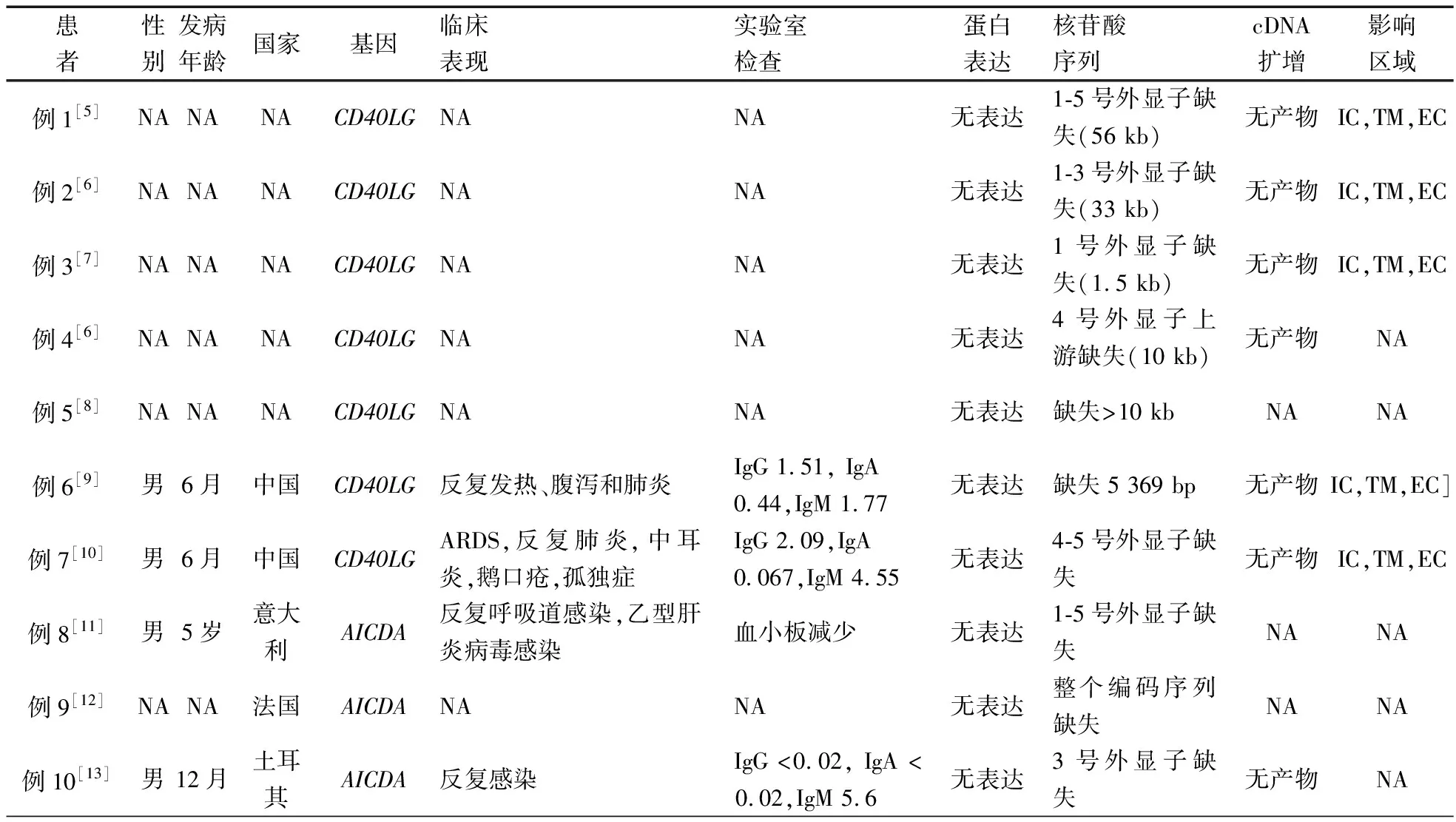
体格检查:T 37℃,HR 115·min-1,R 24·min-1,体重9.5 kg(P3~P10),身高73 cm( 图1 患儿皮肤黄染及腹壁静脉曲张 辅助检查:血常规WBC (9.30~13.16)×109·L-1,N (1.29~5.34)×109·L-1,L (3.62~4.24)×109·L-1,PLT (115~188)×109·L-1,Hb 95~100 g·L-1,ESR 57 mm·h-1,PCT 0.2~27.4 ng·mL-1。体液免疫指标IgG 0.37 g·L-1,IgA 0.04 g·L-1,IgM 1.21 g·L-1,IgE 0.1 IU·mL-1。淋巴细胞免疫分析CD3+80.9%,CD4+54.9%,CD8+23.2%,CD19+15.4%,CD16+CD56+1.96%,总T淋巴细胞计数2 440·μL-1,CD4+T淋巴细胞计数1 751·μL-1,CD8+T淋巴细胞计数740·μL-1,B淋巴细胞计数438·μL-1,NK细胞计数56·μL-1。肝功能AST 98~209 IU·L-1,ALT 138~414 IU·L-1,TBIL 29.6~92.1 μmol·L-1,DBIL 21.0~69.1 μmol·L-1。甲状腺功能FT33.3 pmol·L-1,T30.8 nmol·L-1,FT47.3 pmol·L-1,T499.9 nmol·L-1,甲状腺过氧化物酶抗体(TPOAb)49.8 U·mL-1,甲状腺球蛋白抗体(TgAb)81.1 IU·mL-1。直接Coombs试验可疑,间接Coombs试验阴性。血培养(马尔尼菲青霉菌+);真菌D-葡聚糖420.4 pg·mL-1;血CMV DNA 5.1×102拷贝·mL-1;粪便沙门氏菌核酸检测阳性。骨髓细胞学检测示,骨髓增生明显活跃,成熟阶段中性粒系比例减低,嗜酸性粒细胞增多症(中度)。胸部CT示,两肺散在多发间质性病变,两侧少量胸腔积液(图2A)。上消化道超声:食管下段及胃底静脉迂曲、扩张。上消化道CT平扫+增强+三维重建:肝脏增大,格林生系统增宽,门静脉较细小,第一肝门区分支增多、迂曲,考虑肝门静脉海绵样变性,脾脏体积增大,肝门区及腹膜后软组织样密度影(图2B)。 图2 患儿胸部、腹部CT 免疫分型:表2显示,根据文献报道[1]的方法进行淋巴细胞精细分型(BD公司,美国)。流式细胞仪检测淋巴细胞亚群,结果显示,T、B淋巴细胞数量正常,记忆B细胞及浆细胞明显减少(均为0)。 经患儿监护人知情同意,对患儿及其父母进行应用免疫整体捕获方案(迈基诺,北京)进行全外显子测序,依据1000 Genomes Project,Exome Variant Server(EVS)及Exome Aggregation Consortium(ExAc)数据库,滤除人群中突变频率>1%的变异,去除每个外显子及上下游各50 bp内含子变异、同义变异(除外位于剪切位点的变异)等,同时使用SIFT、PolyPhen等软件对筛选出的突变对蛋白功能的影响进行预测。根据美国遗传学与基因组学学会(ACMG)制定的指南筛选出可能致病的突变[3]。结果显示,全外显子测序点突变分析未见可疑致病突变位点。 拷贝数变异分析:采用cnvkit软件,利用同一pool捕获的样本作对照进行分析。统计target区域和非target区域的reads深度,然后在样本内对深度进行均一化和矫正,和对照集进行比较计算,得到拷贝数的信息,用循环二元分割算法(CBS)对区域进行分割。通过Reads数分析发现患儿CD40LG片段疑似缺失(图3)。 CD40LG基因外显子荧光定量PCR:提取患儿及其父母基因组DNA,根据CD40LG基因的不同外显子设计引物(表2),应用ROCHE480PCR仪,20 μL体系扩增,记录Ct值,采用文献[4]报道的方法进行数据分析。患儿平均Ct值为35,CD40LG基因1~5号外显子大片段缺失。其父亲和母亲平均Ct值分别为26.1和25.8,差异无统计学意义(P>0.05)(图4)。 图3 患者X染色体(Xq25-28)二代测序reads分析结果 图4 患者家系CD40LG基因各外显子qPCR扩增结果 注 P:患儿;PF:患儿父亲;PM:患儿母亲;E1-E5:CD40LG基因DNA1至5号外显子 CD40LG基因cDNA凝胶电泳:使用RNAiso Blood试剂盒(Takara公司,日本)提取RNA,应用PrimeScriptTMII 1st Strand cDNA Synthesis Kit(Takara公司,日本)完成患儿及其父母和正常对照的RNA的反转录。根据CDS序列设计上下游引物(表2),反应体系为25 μL,退火温度为60℃。扩增完成后,对产物进行凝胶电泳。患儿CD40LG基因CDS序列cDNA扩增无产物,其父母cDNA扩增有产物(图5)。 流式细胞术检测淋巴细胞CD40L的表达情况:采集患儿、患儿母亲和健康对照者静脉血0.2 mL,与RPMI-1640(Thermo Fisher公司,美国)培养基混匀,标记为未刺激管;另取抗凝血0.2 mL,与RPMI-1640培养基、PMA(Sigma公司,美国,1 ng·μL-1)、离子霉素(Sigma公司,美国,50 ng·μL-1)作刺激剂,充分混匀,标记为刺激管,孵育4 h。将患儿、患儿母亲和健康对照者的刺激管和未刺激管各分成4管,分别依次加入如下组合抗体(BD 公司,美国):CD3-PerCP/IgG1-PE/CD8-APC,CD3-PerCP/CDl54-PE/CD8-APC,CD3-PerCP/IgG1-PE,CD3-PerCP/CD69-PE。以CD3+CD8-T淋巴细胞代表CD4+T淋巴细胞群作为分析对象,测定CD40L在其表面的表达情况。患儿CD3+CD8-T淋巴细胞CD40L比例为1.0%,其母亲为16.8%,较正常对照明显减少(1.0%,16.8%vs64.7%)(图6)。 图5 患儿家系淋巴细胞CD40LG基因CDS区cDNA扩增后凝胶电泳图 注 A:患儿及对照;B:患儿父母凝胶电泳 治疗与随访:入院后先后予头孢哌酮舒巴坦、米卡芬净、更昔洛韦、伊曲康唑静滴及复方磺胺甲噁唑口服抗感染治疗,住院期间予丙种球蛋白支持治疗及复方甘草酸苷、熊去氧胆酸护肝利胆治疗,加用左甲状腺素片口服治疗,患儿好转出院,出院后定期返院予IVIG治疗,中性粒细胞较前升高(2.8~5.34)×109·L-1,仍间断反复感染,皮肤黄染等肝功能损害较前好转(18月龄复查ALT 49 IU·L-1,AST 70 IU·L-1),生长发育仍受限。 检索PubMed、中国知网、万方和维普数据库,检索时间均从建库至2019年4月11日。英文检索关键词“Hyper IgM syndrome”,共检出3 893篇;中文检索关键词“高IgM综合征”,共检出233篇。HGMD数据库共收录HIgM综合征致病突变295个,CD40LG最多见(234个)。常见致病突变类型为错义突变、无义突变、缺失突变、剪切位点突变,其中以错义突变最多见。AICDA、CD40、UNG、IKBKG基因较CD40LG报道相对较少,亦以错义突变最常见。拷贝数变异导致的HIgM综合征病例的相关文献共10篇(n=10),其中英文8例,中文2例。7例为CD40LG基因拷贝数变异,均表现为大片段缺失,2例为国内报道(不含本文报道,P6和P7);3例为AICDA基因拷贝数变异,亦为大片段缺失,余致病基因无拷贝数变异(表3)。 表3 CNV所致HIgM综合征病例汇总 注 IgG、IgA和IgM单位g·L-1;IC:胞内区,TM:胞浆区,EC:跨膜区 原发性免疫缺陷病(PID)是单基因突变导致免疫器官、免疫细胞及免疫活性分子发生缺陷,最终导致机体免疫功能异常的一组临床综合征。PIDs常缺乏明显的基因型与表型相关性,同一基因不同突变可导致不同的临床与免疫表型(RAG1基因不同突变可导致SCID、OMENN综合征等), 而不同的基因突变又可以导致相似的临床表型(OMENN综合征可由RAG1、RAG2、LIG4、ADA等基因突变导致)。近年来每年都有很多新的PID致病基因通过NGS被确认。但即使在二代测序技术的帮助下,最高也仅有30%~40%的PID患者得到基因诊断,在二代测序阴性病例中,拷贝数变异(CNVs)越来越受到关注。 CNV是由基因组重排导致的,一般指的是长度>1 kb的大片段拷贝数增加或减少,主要表现为亚显微水平的缺失和重复[14]。随着测序技术的广泛应用,CNV在PID的作用逐渐受到重视,包含DOCK8、CYBB、BTK等在内的数十种PID相关基因已有CNV的相关报道[15]。目前全世界仅7例CD40LG基因大片段缺失报道,加上本文报道共8例,及3例AICDA基因CNV报道,本文为首次系统整理CNV所致高IgM综合征(HIgM)。 HIgM为免疫球蛋白类别转换障碍伴或不伴体细胞高频突变的一种原发性免疫缺陷病,典型的免疫学表现为血IgG、IgA水平下降或缺乏,血IgM水平正常或升高。临床主要表现为反复感染,如肺部细菌感染、中耳炎、胃肠道感染等,某些机会致病性感染发生率高,如卡式肺囊虫、隐孢子虫、弓形虫感染等,且易罹患某些自身免疫性疾病及恶性肿瘤。经典的HIgM综合征分为5类:①CD40L缺陷;②CD40缺陷;③活化诱导的胞苷脱氨酶(AID) 缺陷;④尿嘧啶DNA转葡糖基酶(UNG)缺陷;⑤其他,仅根据表型难以完全明确致病分子。 CD40L属肿瘤坏死因子超家族成员,表达于活化的CD4+T细胞表面,CD40属TNF受体家族成员,持续表达于B细胞及单核巨噬细胞、树突状细胞等抗原提呈细胞(APC)[16]。CD40-CD40L共刺激信号促进B细胞活化、类别转换及体细胞高频突变,诱导记忆B细胞的形成,从而使B细胞产生的抗体由IgM向IgG、IgA或者IgE转换并且对T细胞依赖性抗原产生高亲和力的抗体[17],同时影响CD4+T细胞和树突状细胞及巨噬细胞的相互作用,使细胞免疫受损。因此,CD40L的缺陷将导致导致X连锁HIGM患者IgG、IgE和IgA水平明显下降,但IgM水平正常或升高;XHIGM患者存在正常数量的成熟B细胞,但是不能转换为记忆B细胞,记忆B细胞数量减少。 本文患儿以反复感染起病,病程中出现马尔尼菲青霉菌、鼠伤寒沙门菌等机会致病菌感染,伴中性粒细胞减低及自身免疫性溶血性贫血、桥本甲状腺炎等自身免疫疾病,外周血IgM水平正常,IgG、IgE和IgA均降低,逐渐出现肝胆系统病变,记忆B细胞及浆细胞明显减少,临床符合HIgM表现,由于缺乏阳性家族史,难以明确致病分子。据HGMG数据库,目前HIgM综合征致病突变中,最常见的突变类型是点突变,其中错义突变最为多见。本文患儿二代测序常规生物信息学分析并未见致病突变位点,但患儿临床表型典型,流式细胞术检测患儿CD3+CD8-T淋巴细胞CD40L蛋白亦无表达,遂进一步行CNV分析,发现CD40LG基因大片段缺失,进一步扩增其cDNA无产物,从而明确CD40LG基因CNV所致HIgM综合征。 目前HIgM的基因型与临床表型的关系尚不明确,但本文患儿大片段缺失与点突变患者临床表型无明显差异。例7除HIgM综合征常见临床表现外还存在孤独症,因曾有报道提示孤独症谱系障碍(ASD)患者[18]中有高频率新发生的CNV,尤其是缺失型CNV,推测其独特表型可能与CNV影响除CD40LG以外的其他基因有关。本文患儿母亲为杂合CNV携带者,其CD40L表达降低,与点突变的携带者相比,虽无明显感染表现,但其CD40L表达水平相对低下,因此母系家族中CD40L表达降低可能是作为大片段缺失变异的高危因素。 致谢:感谢迈基诺有限公司史丽娜老师对此文的帮助与指导。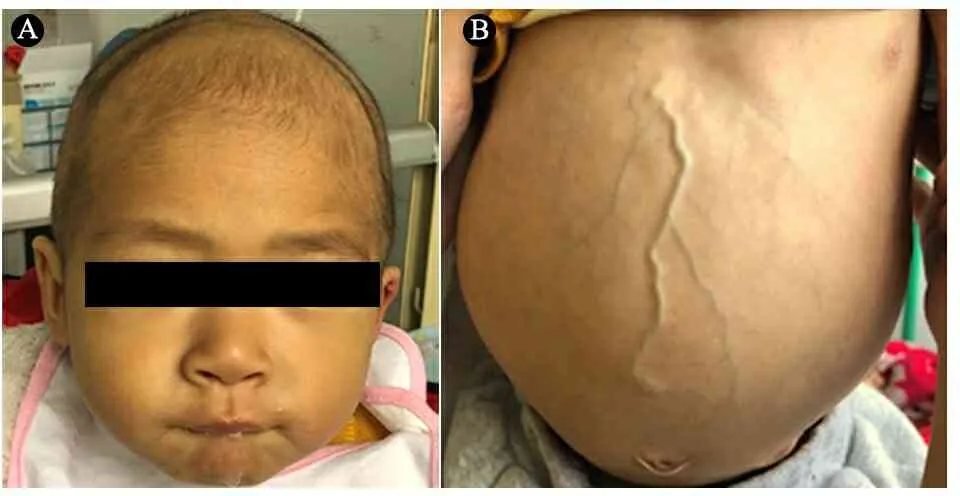
2 文献复习
3 讨论
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
中国食物与营养(2022年5期)2022-06-17
右江民族医学院学报(2022年2期)2022-05-19
河北医学(2021年10期)2021-10-27
青岛大学学报(医学版)(2021年4期)2021-09-10
中国农业科学(2021年6期)2021-03-25
中国生殖健康(2020年4期)2021-01-18
当代畜禽养殖业(2020年9期)2020-10-21
家庭医学·下半月(2020年2期)2020-04-26
科学生活(2019年7期)2020-01-01
