
通过抑制ATM上调Bim促进小脑颗粒神经元凋亡
2019-05-30 06:36刘锶锶曾淑莲吴力强黄紫燕王业忠袁忠民
中山大学学报(医学科学版) 2019年3期
刘锶锶,曾淑莲,吴力强,黄紫燕,郅 程,王业忠,袁忠民
(广州医科大学附属第二医院神经外科//神经外科疾病研究中心,广东 广州 510260)
凋亡是程序性死亡的主要重要形式,其与细胞增殖共同维持机体正常生长发育和内环境的稳定[1]。神经退行性疾病是由神经元及其髓鞘丧失而出现功能障碍的一类神经系统疾病,包括阿尔兹海默症、帕金森病、肌萎缩性脊髓侧索硬化症等[2]。这些疾病的共同的病理改变是大量细胞凋亡引起脑内神经元减少[3]。细胞凋亡涉及一系列基因的调控及蛋白的合成,并受促凋亡基因的活化调节。DNA损伤、线粒体膜流动性降低等均可启动线粒体依赖的凋亡通路,导致相关蛋白的释放。BH3-only家族蛋白Bim上调是神经元发生凋亡的关键事件,其表达上调可以促使神经元发生线粒体依赖的凋亡[4-5]。共济失调毛细血管扩张突变(ataxia telangiectasia mutated,ATM)蛋白是一种丝/苏氨酸蛋白激酶[6],在DNA损伤修复中扮演着重要的角色。目前研究发现,DNA损伤后通过活化ATM-p38MAPKMK2通路,激活下游底物p53,从转录水平上调节基因表达,导致细胞凋亡[7],表明ATM在细胞凋亡的调控中的重要作用,但在神经元凋亡过程中ATM的作用尚不明确。我们想要探索ATM调控神经元凋亡中的机制,并发现其与下游的凋亡蛋白Bim之间的调控关系,从而证实ATM对神经元的存活具有重要意义。希望通过本研究为确立以ATM和Bim为靶治疗DNA损伤引起的神经元凋亡及相关神经病变提供理论支持和实验依据。本研究以SD乳鼠小脑颗粒神经元作为研究对象建立体外神经元存活和凋亡模型,探讨ATM在小脑颗粒神经元凋亡过程中的作用与机制。
1 材料与方法
1.1 实验材料
出生7~8 d SD乳鼠,性别不限,购于中山大学实验动物中心。Basal Medium Eagle(BME)和Fetal bovine serum购于美国Gibco公司。钙磷转染试剂盒购于美国Promega公司。细胞实验所用24孔板和6孔板购自美国Corning公司。ECL发光试剂和Hoechst 33258荧光染料购于美国Sigma公司。KU-55933购于Selleck公司。Anti-ATM、Anti-p-ATM、Anti-Bim、Anti-Caspase 3、Anti-GAPDH购自美国CST公司。
1.2 方法
1.2.1 神经元的原代培养 小脑颗粒神经元来自7~8 d SD乳鼠(15~19 g)。从健康的乳鼠脑组织中分离出小脑组织,机械破碎后用胰酶以及DNA酶处理得到单细胞悬液,以1.5×106cells/mL的密度接种于细胞板中,置于37℃、体积分数为5%CO2及饱和湿度条件下培养。培养基为含10%FBS的BME,以及 25 mmol/L KCl[8]。接种后 24 h 添加阿糖胞苷(10 μmol/L)限制胶质细胞的生长。细胞在体外培养7 d后,用含钾离子浓度为25 mmol/L或5 mmol/L的无血清培养基替换原培养基,实验组用 KU-55933(10 μmol/L)处理细胞,不添加抑制剂的细胞组加入DMSO作为对照。
1.2.2 免疫印迹法 直接加入含DTT的上样缓冲液收集细胞,超声,变性、上样、电泳、转膜、5%脱脂牛奶室温封闭1 h后,分别加入一抗(1∶1 000):p-ATM、ATM、Bim、Caspase 3、GAPDH、β-tubulin,4℃摇晃过夜,第2天用TBST洗膜10 min×3次,再分别加入对应种属的二抗(1∶5 000),室温孵育1 h,TBST洗膜10 min×3次后行ECL发光,应用胶片曝光。
1.2.3 凋亡测定 处理小脑颗粒神经元后,加入Hoechst 33258(1∶200)染料,置于37 ℃培养箱染色1 h后,于荧光倒置显微镜下观察并拍照。通过相对于同一视野中核固缩的细胞数与同一视野中细胞总数的比值来表示神经元凋亡率。
1.3 统计学方法
采用SPSS19.0统计软件进行统计学处理,计量资料采用均数±标准差()或四分位数P50(P25,P75)表示。首先对样本进行正态性检验和方差齐性检验,对符合正态分布并且方差齐性的数据,两样本均数的比较采用两独立样本t检验;3组及以上样本的均数比较采用单因素方差分析,随后的组间比较采用LSD法;否则3组及以上样本的均数比较采用H检验;P<0.05被认为差异有统计学意义。
2 结果
2.1 在体外神经元凋亡(5K)条件下,ATM及其磷酸化水平降低
为了研究ATM在小脑颗粒神经元凋亡中的作用,我们首先利用25和5 mmol/L KCl在体外建立神经元存活(25 K组)和凋亡(5 K组)模型。Caspase 3是凋亡特异性标志物,其活化型(Cleaved-caspase 3)的表达水平可以反映细胞凋亡情况,而Bim上调是诱导神经元发生线粒体依赖凋亡及Caspase 3激活的关键事件。Western Blot实验结果显示,与25 K组比较,5 K组Cleavedcaspase 3表达水平升高(P<0.05),同时Bim表达也增加(P<0.05,图1),说明成功建立体外神经元凋亡模型。此外与25 K组比较,5 K组的p-ATM和ATM的表达水平下降(P<0.05;图1)。
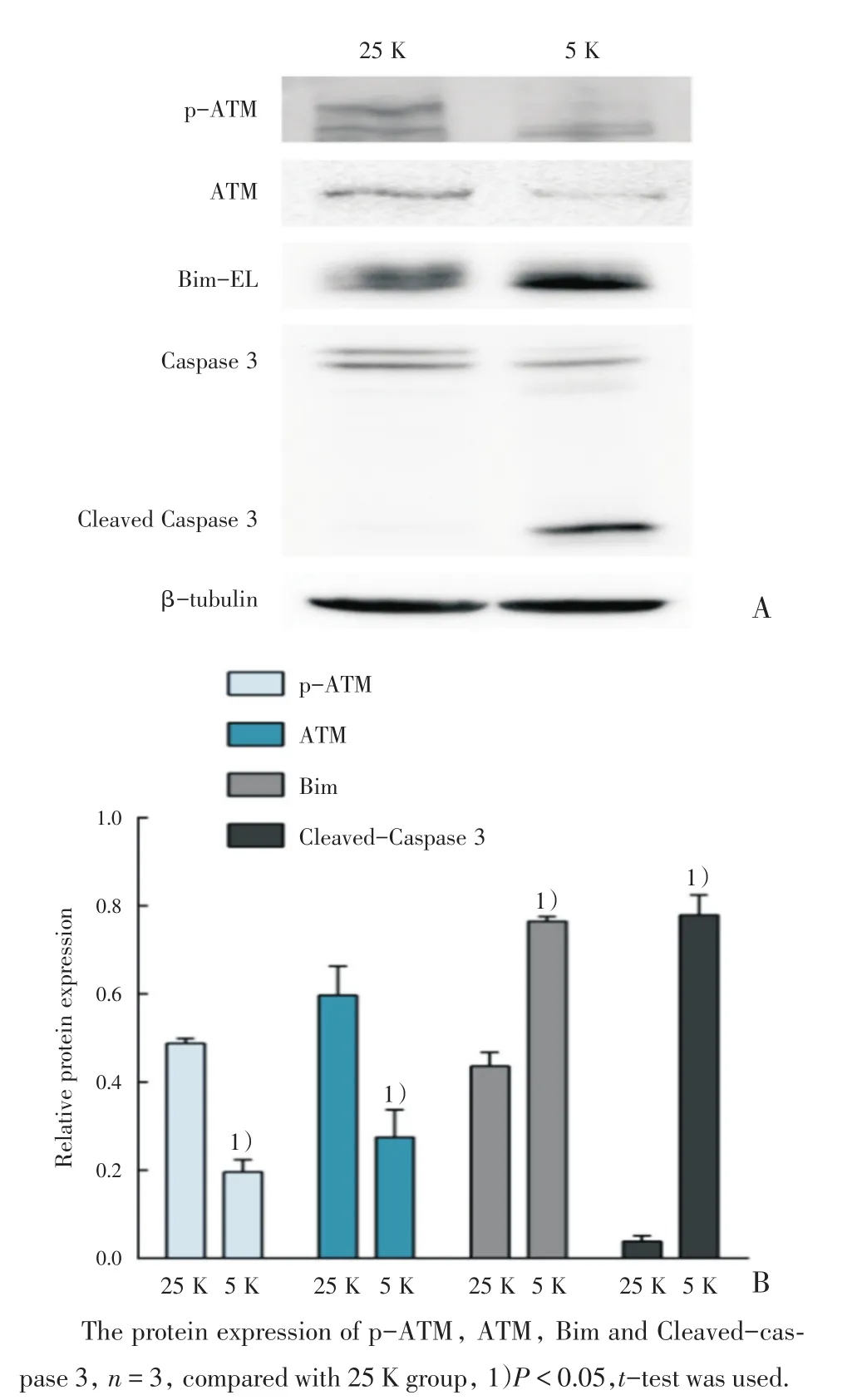
图1 5 K条件下小脑颗粒神经元ATM及其磷酸化水平降低Fig.1 5K caused a significant reduction in ATM and p-ATM in CGN
2.2 在25 K条件下,抑制ATM导致Bim表达水平增加,Caspase 3激活
在25 K条件下,我们利用KU-55933抑制ATM活性,与25 K组比较,ATM抑制组中p-ATM、ATM的表达均降低(P<0.05;图2B),说明KU-55933使p-ATM和ATM的表达水平下降。同时Bim、Cleaved-caspase 3的表达水平均升高(P<0.05;图2C),提示抑制ATM能够使小脑颗粒神经元发生线粒体依赖的凋亡。
2.3 在25 K条件下,抑制ATM增加神经元核固缩率
随后在25 K条件下,我们利用KU-55933抑制ATM后进行Hoechst染核,观察小脑颗粒神经元的核固缩情况。与25 K组比较,5 K组和ATM抑制组的凋亡率均增加(P<0.05;图3)。
2.4 抑制Bim将降低ATM抑制剂引起的神经元凋亡率
为进一步探讨抑制ATM是否导致Bim依赖的小脑颗粒神经元凋亡,在抑制ATM条件下,分别用光辉霉素A(Mithramycin A,MMA)和色霉素A3(Chromomycin A3,CMA3)处理细胞,检测其对Bim和Caspase 3表达的影响[9]。在ATM抑制组中,与对照相比Bim的表达增加(P<0.05;图4A、B)且Cleaved-caspase 3表达增加(P<0.05)。而MMA和CMA3能显著抑制KU-55933诱导的Bim表达和Cleaved-caspase 3活性(P <0.05;图4B、C)。
接下来,我们同时抑制Bim和ATM后进行Hoechst染核色,观察小脑颗粒神经元的核固缩情况。与ATM抑制组比较,MMA组、CMA3组中小脑颗粒神经元核固缩率降低,差异具有统计学意义(P < 0.05;图4D、E)。
3 讨论
凋亡是许多生理和病理过程中细胞发生程序性死亡的主要形式,一直以来都是科学研究热点。而神经元的凋亡被认为是中枢神经系统发育的正常特征,并在神经退行性疾病和衰老中有重要的作用。例如在神经退行性疾病中,阿尔茨海默病的特征性病理变化之一是脑内神经元数目明显减少,而帕金森病的主要病理特征是多巴胺能神经元的减少[10]。在脑内神经元凋亡受到一系列基因的调控以及蛋白表达的影响,包括促凋亡基因的活化调节。其中,经典的线粒体依赖的凋亡通路是,当细胞受到各种因素刺激后,细胞色素C从线粒体中释放,与凋亡活化因子-1(Apaf-1)、pro-Caspase 9形成凋亡体,激活Caspase 9,继而激活下游Caspase 3,引起细胞凋亡[11]。有许多因素可能引发神经细胞凋亡,低钾(low potassium,也有文献称撤钾,potassium deprivation,本文称5 K)是其中一种。在CGN的培养中,可以通过去极化细胞外钾离子(25 mmol/L KCl,25 K)的浓度来维持CGN存活,当钾离子浓度降低至5 mmol/L KCl(撤钾)时会引发典型的细胞凋亡[12],该模型被广泛用于研究神经元凋亡的机制[9]。我们在5 K条件下检测到Caspase 3的激活,成功建立体外神经元凋亡模型。在此模型上,我们发现在5 K的条件下ATM的表达水平降低。ATM是一种丝/苏氨酸蛋白激酶,在协调DNA损伤反应中起重要作用,包括细胞周期检查点控制,DNA修复和细胞凋亡[13]。当细胞发生DNA损伤时,胞内识别DNA双链断裂(DNA double-strand breaks,DSB),ATM蛋白无活性的二聚体分子内发生自身磷酸化,解离成有活性的ATM蛋白单体,进而诱发下游相应底物发生一系列的磷酸化级联反应,最终引起细胞周期阻滞、DNA修复或细胞凋亡[14]。我们的研究发现,在神经元中抑制ATM后能上调Bim的表达,激活了Caspase 3,导致神经元的凋亡,提示ATM在调控细胞凋亡发挥重要的作用。
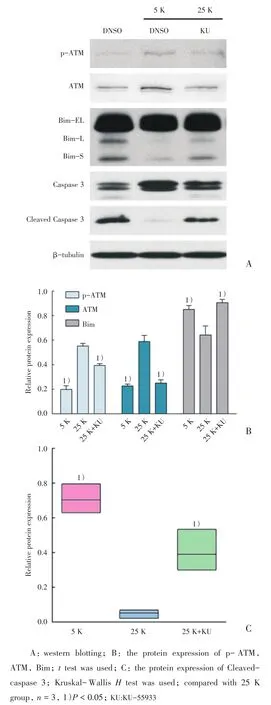
图2 25 K条件下抑制ATM增加Bim和Caspase 3的表达Fig.2 Inhibition of ATM induced Bim expression and Caspase 3 activationat 25 K
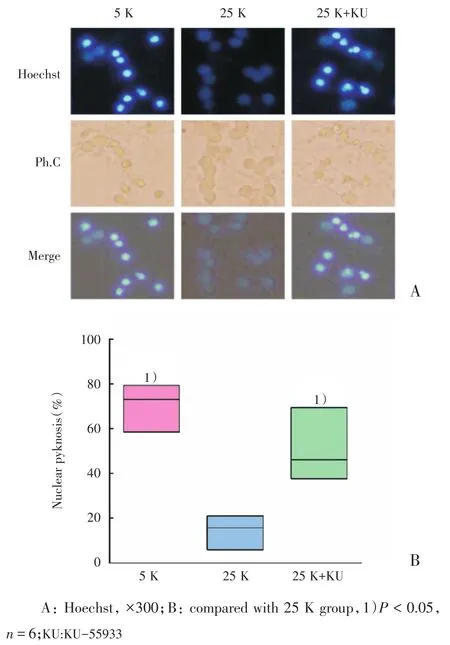
图3 抑制ATM增加小脑颗粒神经元核固缩率Fig.3 ATM inhibition increased the nuclear pyknosis rate of cerebellar granule neurons
Bim是Bcl-2家族成员之一,属于促凋亡的BH3亚家族。它只含有一个BH3同源结构域,表达上调可以促进线粒体依赖的细胞凋亡[4-5]。研究报道,BH3-only家族蛋白Bim上调是神经元发生凋亡的关键事件[4]。而Bim的表达上调是由上游活性分子进行调控的,研究发现抑制CaMK II能上调Bim的表达进而引起神经元凋亡[15]。同时有研究报道[9],GCN5的表达下调也能够诱导Bim依赖的神经元凋亡。而本研究发现,抑制ATM后Bim的表达水平升高,进而激活Caspase 3,引起神经元凋亡,说明ATM通过调控Bim的表达从而发挥调控细胞凋亡的作用。Wu等[9]报道Egr-1通过转录水平调控Bim的表达从而调节细胞凋亡。我们同时抑制Egr-1的转录活性和ATM后发现,Bim的表达水平显著减少,神经元的凋亡率也随之降低,说明抑制ATM可能通过上调Egr-1的转录活性从而上调Bim的表达,导致小脑颗粒神经元凋亡。
总之,本研究发现在小脑颗粒神经元中,ATM活性通过抑制Bim表达促进细胞存活,而ATM表达减少会使促凋亡蛋白Bim的表达增加,激活Caspase 3,进而导致神经元发生凋亡。据我们所知,这是ATM调控神经元存活和凋亡的新机制。本研究为确立以ATM和Bim为靶治疗DNA损伤引起的神经元凋亡及相关神经病变提供理论支持和实验依据。ATM调控Bim的机制仍需进一步研究。
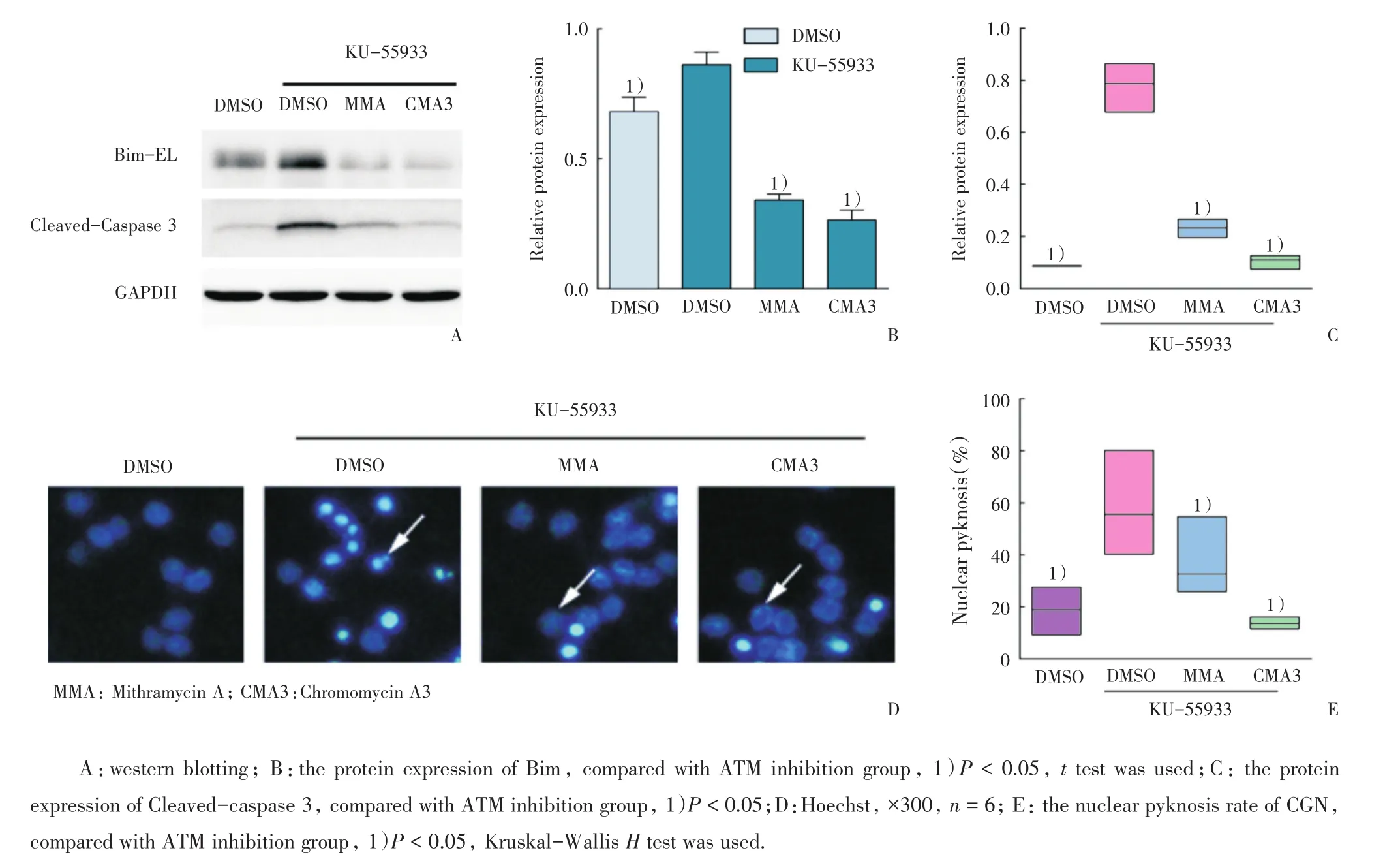
图4 抑制Bim降低ATM抑制剂引起的小脑颗粒神经元凋亡Fig.4 Inhibition of Bim by Mithramycin A or Chromomycin A3 decreased the apoptotic rate of cerebellar granule neurons induced by ATM inhibitor
猜你喜欢
医学研究生学报(2022年5期)2022-12-07
中国医药导报(2022年28期)2022-11-25
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中国典型病例大全(2022年12期)2022-05-13
中国卒中杂志(2021年7期)2021-11-29
电子产品世界(2021年8期)2021-01-16
好孩子画报(2019年8期)2019-09-19
中国计算机报(2019年49期)2019-02-07
中国疼痛医学杂志(2019年9期)2019-01-04
哲思2.0(2017年12期)2017-03-13
