
高效制备BDD单体新方法探究
2019-05-28 02:12张鹏兵高文婷刘晨旭周鹏鑫
云南化工 2019年3期
张鹏兵,高文婷,刘晨旭,周鹏鑫
(西北师范大学化学化工学院,甘肃 兰州 730070)
BDD,化学名称为1,3-双 (5-溴噻吩基)-5,7-双异辛基苯并 [1,2-c:4,5-c']二噻吩-4,8-二酮,英文名称:1,3-bis(5-bromothiophen-2-yl)-5,7-bis(2-ethylhexyl)benzo[1,2-c:4,5-c']dithiophene-4,8-dione,化学结构如图1所示,是一种重要的有机合成中间体,在合成共轭聚合物给体材料中具有重要应用[1-15]。
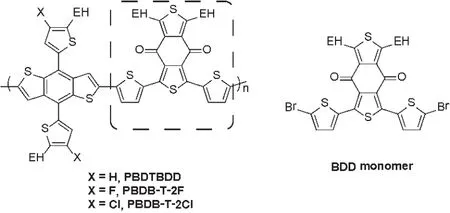
图1 BDD单体及其衍生共轭聚合物结构Fig 1 Chemical structure of BDD and its derivative conjugated copolymer.
聚合物PBDTBDD最早是由中科院化学所侯剑辉课题组报道的一类共轭聚合物给体材料,PBDTBDD是一类窄带隙吸收材料,吸收光谱范围在500~700 nm,在溶液中具有很强的聚集效应[1]。由于该材料具有优异的光谱吸收性能,该材料及其衍生物广泛应用于有机太阳能电池活性层材料中。该材料作为电子给体与PC61BM作为电子受体组装的有机太阳能电池的光电转换效率可以达到6.67%。目前,由该材料作为电子受体组装的三元有机太阳能电池光电转换效率突破10%[15]。含有该结构单元的聚合物给体材料可以实现超过14%的光电转化效率[6,7]。南开大学陈永胜课题组今年在Science上报道了目前有机太阳能电池光电转化效率的世界最高纪录17.3%,该器件的活性层材料中含也含有BDD结构单元[16]。文献调研发现,BDD结构单元是高效率太阳能电池活性层材料的重要结构单元,含有BDD单元的活性层材料引领着有机太阳能电池的光电转化效率。因此,对BDD单体的合成工艺进行研究具有非常重要的意义。合成路线见图2。
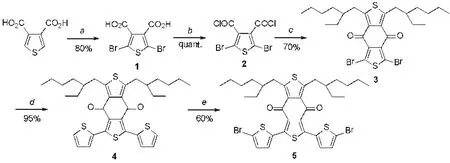
图2 BDD单体合成路线Fig.2 Synthetic route of BDD monomer.
能源偿还时间(EPBT)是衡量太阳能电池使用过程中产生的能量补偿制造过程中消耗能量所需的时间[16]。提高和改进聚合物单体的合成效率,不仅可以降低聚合物材料的合成成本,而且缩短EPBT。BDD单体现有合成方法是从3,4-噻吩二羧酸出发,经溴化,酰氯化再与2,5-二异辛基噻吩发生双傅克酰基化反应关环生成中间体3。化合物3与噻吩-2-三丁基锡经钯催化的Stille偶联反应生成化合物4,最后用NBS在DMF中双溴化生成BDD单体(Scheme 1)[12]。最后一步使用高沸点DMF作为溶剂,室温反应3h,产率中等(60%),制约了BDD单体的合成效率。此外,该反应后处理一般需要经过萃取分液、洗涤、干燥、过滤、浓缩,操作繁琐。本文对该过程条件进行了详细考察,改进后处理过程,将分离产率提高至90%以上。本方法极大地提高了步骤经济性、减少溶剂用量,使后处理过程更为绿色环保。
1 材料及方法
1.1 仪器与试剂
1H NMR和13C NMR在Bruker arx-400核磁测试仪上进行,CDCl3或 (CD3)2SO为溶剂;TMS为内标。噻吩-3,4-二羧酸,2-三丁基甲锡烷基噻吩,草酰氯,2,5-双异辛基噻吩,氮溴丁二酰亚胺 (NBS)均购自国药控股化学试剂有限公司。催化剂AlCl3和Pd(Ph3)4购自Alfa Aesar试剂公司。NBS用水重结晶后使用。四氢呋喃、乙腈、二氯甲烷、二氯乙烷、氯仿等均为分析纯试剂,购自上海泰坦科技有限公司;TLC和柱层析用青岛海洋化工厂生产GF254硅胶板和柱层析硅胶(200~300 目)。
1.2 化合物1的合成[1]
噻吩-3,4-二羧酸(5.0 g,29.1 mmol)置于100 mL单口烧瓶中,加入50 mL冰醋酸,恒压滴液漏斗滴加液溴(10.2 g,64.0 mmol),0.5h滴加完毕。(注意:尾气用碱液吸收)。将反应混合物在室温搅拌过夜。反应毕,将反应混合物倒入100mL碎冰中,析出大量淡黄色固体,搅拌10min,抽滤,用冰水洗涤三次,干燥,得7.7 g白色固体,产率80%。MS:m/z=330.13C NMR(d6-DMSO,100 MHz),δ:162.6,135.2,114.4.
1.3 化合物2的合成[1]
2,5-二溴噻吩-3,4-二羧酸(2.0 g,6.0 mmol)置于25 mL单口烧瓶中,分别依次滴加10 mL干燥二氯甲烷,1滴DMF,然后缓慢滴加草酰氯(2.0 mL,24 mmol)。反应体系由浑浊变澄清,反应12h。然后将过量的草酰氯和二氯甲烷旋除,直接用于下一步反应。
1.4 化合物3的合成[1]
在上述反应体系中,加入10mL 1,2-二氯乙烷和2,5-双异辛基噻吩(1.9 g,6.0 mmol),冰浴冷却至0℃,分批加入无水三氯化铝(6.4 g,48 mmol),在0℃下搅拌0.5h,再在室温搅拌3~5h。将反应液倒入30 ml 1mol/L的冰盐酸中,用氯仿萃取3次(30 ml/次),合并有机相,无水硫酸镁干燥,过滤,旋除溶剂。柱色谱分离(PE/DCM=5∶1),分离得到2.6 g淡黄色固体,产率70%。1 H NMR (400 MHz,CDCl3),δ:3.83-3.03 (m,2 H),1.75 (dd,J=12.2,6.0 Hz,1H),1.48-1.19(m,8H),0.89 (dt,J=11.2,7.2 Hz,6H).13C NMR(CDCl3,100 MHz),δ:175.5,155.3,134.9,132.8,119.5,41.2,34.0,32.7,28.8,26.0,23.1,14.2,10.9。
1.5 化合物4的合成[1]
在100mL Schlenk反应瓶中称取化合物4(1.5 g,2.5 mmol),加入20毫升甲苯,噻吩三丁基锡(2.8 g,7.5 mmol)和四三苯基膦钯(60 mg,1 mol%),用高纯氩气置换空气5次,在110℃反应16 h。TLC监测反应完毕,将反应体系冷却,旋除甲苯,柱色谱分离(PE/DCM=5∶1),得黄色固体1.5 g,产率99%。1H NMR (400 MHz,CDCl3)δ:7.74 (d,J=4.0 Hz,2H),7.49 (d,J=4.0 Hz,2H),7.16-7.09 (m,2H),3.72-2.99(m,4H),1.76 (dd,J=12.0 Hz,6.0 Hz,2H),1.52-1.21 (m,16H),1.02-0.80 (m,12H).13C NMR(100 MHz,CDCl3),δ:177.6,153.5,142.4,133.4,133.1,132.6,130.5,129.3,127.1,41.2,33.6,32.7,28.8,26.0,23.0,14.1,10.9。
1.6 化合物5(BDD单体)的合成
将化合物5(12 mg,0.02 mmol)和NBS(9.0 mg,0.05 mmol)置于5 mL试管中,加入1.0 ml二氯甲烷。室温反应1 h。将反应体系拌硅胶,直接柱色谱分离(PE/DCM=8∶),得黄色固体13 mg,产率90%。1H NMR (400 MHz,CDCl3)δ:7.42(d,J=4.0 Hz,2H),7.05(d,J=4.0 Hz,2H),3.35-3.24 (m,4H),1.76-1.72 (m,2H),1.41-1.32 (m,16H),0.94-0.90 (m,12H).13C NMR (100 MHz,CDCl3)δ:177.1,153.8,141.2,134.6,132.5,131.5,130.1,129.5,118.4,41.1,33.7,32.9,29.0,26.1,23.1,14.3,10.9。
2 结果与讨论
通过实验对比发现,制约BDD的效率的根本原因在于化合物4与NBS的溴化反应(图3),为了提高该反应的产率,我们对影响反应的各种因素诸如溶剂、浓度、温度、底物当量比以及加料方式等进行优化。
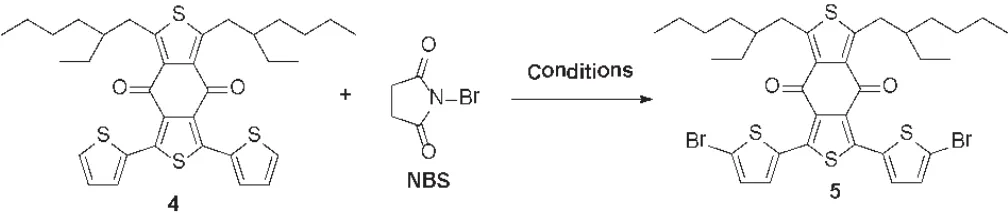
图3 NBS溴化反应过程Fig.3 Process of NBS bromination reaction
2.1 溶剂对反应的影响
已报道的研究中,该类溴化反应主要使用高沸点溶剂DMF作为溶剂,室温反应3 h,BDD单体最高分离产率仅为60%。为此,实验分别考查了甲醇、二氯甲烷、三氯甲烷、乙酸乙酯、石油醚、四氢呋喃、二甲基亚砜、N,N-二甲基甲酰胺、水等作为反应溶剂。实验表明(见图4),该反应在水中几乎不能进行反应,但在其他溶剂中均可反应,产率相差较大。石油醚用作溶剂时,由于底物在石油醚中的溶解度低,导致反应产率BDD产率小于5%。在极性非质子溶剂DMF,DMSO中,BDD产率仅为34%和5%。在质子性溶剂甲醇作溶剂的条件下,该反应能很好的进行,产率可以达到84%,但是在甲醇/水中条件下,BDD产率则降低至44%,这可能与反应物的溶解性有关。以溶剂乙酸乙酯中、氯仿、四氢呋喃、二氯甲烷作为反应溶剂,BDD产率分别提高至82%、81%、90%和91%。基于效果、成本、环保及后处理的因素,我们选择二氯甲烷作为该反应的最佳溶剂。
2.2 浓度对反应的影响
合适的反应浓度既能保证反应顺利进行,又可以减少副反应的发生。我们对底物4的反应浓度(0.01~0.10mol/L)进行了考察。实验表明:随着底物浓度的增加,反应速率呈明显加快,反应时间明显缩短(见图5)。当底物浓度从0.01mol/L增加到0.04mol/L时,反应时间从120min完成减小至30min即可完成,以高达91%的分离产率得到目标产物。当底物浓度增加到0.08mol/L时,反应只需20min即可反应完毕,然而产率从91%下降至80%;当进一步增加底物浓度,产率下降至75%。这是由于随着溶剂用量的进一步减少,虽然底物浓度增加,原料却不能完全溶解,不利于反应物之间充分接触,反应效果反而变差。结果表明:0.02~0.08 mol/L是该反应的较优浓度范围,最终我们选择0.04 mol/L作为该反应的最佳浓度,进一步考察其它因素对该溴代反应的影响。
2.3 反应温度及其它因素反应的影响
降低反应温度至0℃,可以明显观察到反应速率的降低,反应时间变长。在反应过程中用TLC监测反应,能够监测到反应中间体单溴代BDD的生成。将反应温度升高至二氯甲烷回流温度,发现多溴代产物增加,不利于目标产物生成。增加NBS的用量可以提高反应产率,使用2.5当量的NBS即可使底物和中间体全部转化为产物。加料顺序对该反应没有明显影响。延长反应时间至2~12h,反应产率没有明显变化。
2.4 后处理改进
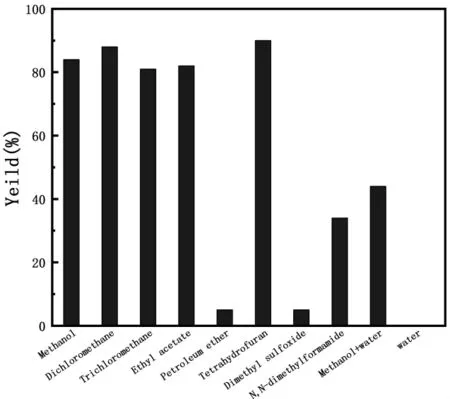
图4 溶剂对反应的影响(化合物4,0.02mmol;NBS 0.04 mmol;溶剂 1.0 mL;反应 3h)Figure 4 Solvent effect on the reaction (Compund 4,0.02mmol and NBS,0.04 mmol,time,3 h,solvent volume,1mL).
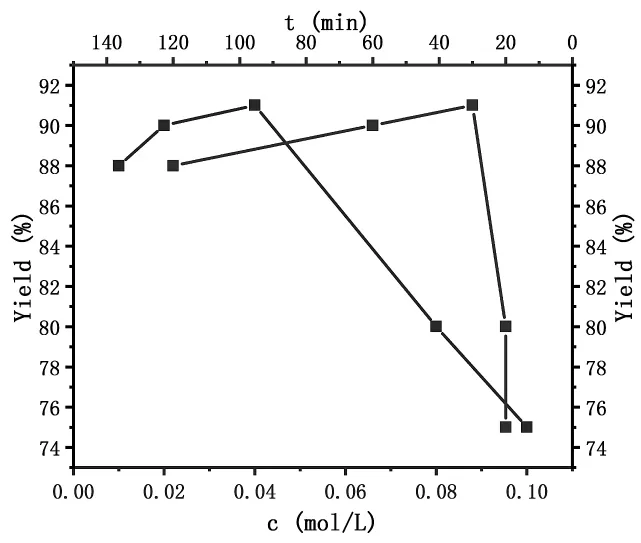
图5 浓度、时间和对反应产率的影响(25℃;化合物4,0.02mmol;NBS,0.04 mmol).Figure 5 Concentration effect on the reaction at 25℃ (Compund 4,0.02mmol and NBS,0.04 mmol).
我们不仅对反应条件进行了优化,而且对反应的处理过程也进行了改进。文献中由于使用到高沸点DMF作为溶剂,后处理采用传统的后处理方式,向反应体系中加水淬灭反应,用氯仿萃取三次,水洗,无水硫酸镁干燥,过滤,浓缩,硅胶拌样,柱色谱(PE/DCM=5∶1) 分离得产物(图4)。本方法由于反应体系非常干净,且使用低沸点溶剂,直接用硅胶拌样,即可以柱色谱分离。避免了萃取分液、洗涤、干燥、过滤、浓缩等操作单元,提高了步骤经济性,节省了溶剂的使用,使得后处理过程更为绿色环保。
2.5 反应机理推测
经实验优化可知该反应溴代发生在噻吩环的α-位上,属于芳杂环上的亲电取代反应。自由基反应通常要求无水无氧条件,需要自由基引发剂(BPO或AIBN)或者光照、加热等自由基引发条件,反应一般发生在烯丙位或苄位上。该反应溶剂无需特殊处理,无需氮气保护,在室温条件下即可反应。溴代反应发生在噻吩环的α-位上,甚至在质子性溶剂(甲醇)中也能进行反应。因此,推测该反应是按照芳环上的亲电取代反应机理进行,NBS作为溴源(图6)。为了验证该推测机理,在反应体系中加入一滴氢溴酸水溶液,观察到反应液立即变为红棕色,继而褪色,TLC监测反应显示反应结束。氢溴酸可以加速该反应的进行。另外,在反应体系中也可以分离得到白色的琥珀酰亚胺,进一步证明了该历程。
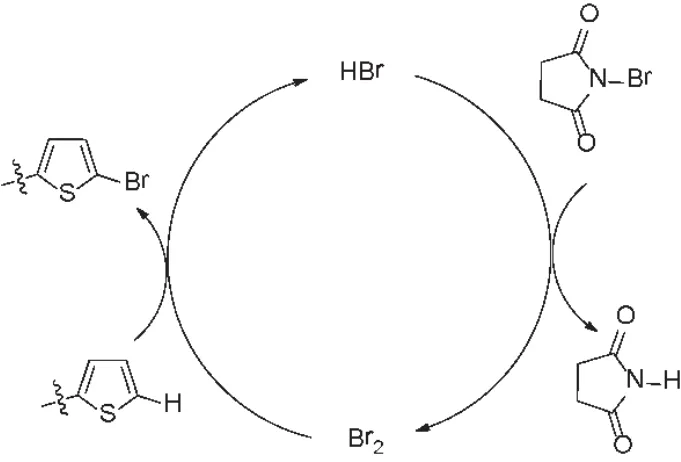
图6 溴化反应机理推测Figure 6 Proposed mechanism of the bromination
3 结论
通过对溴化反应溶剂、浓度、温度及底物当量比研究发现二氯甲烷为溶剂条件下,底物浓度为0.04 mol/L时,仅使用2.0倍当量的NBS在室温反应30min,BDD单体合成中的溴化反应产率由原来60%快速提高至90%以上,得到高质量的BDD单体。同时,我们也对该反应的后处理过程进行了改进。将文献中的后处理操作由原来的7个单元操作缩短为2个,极大地简化了操作步骤,使得后处理操作过程简单易行,极大程度上降低聚合物材料成本。同时,有效地缩短有机太阳能电池器件的能源补偿时间,应用前景广阔。
猜你喜欢
煤炭学报(2022年11期)2023-01-07
化学工程师(2022年3期)2022-04-19
上海化工(2021年2期)2021-04-23
科学(2020年2期)2020-08-24
石油与天然气化工(2020年1期)2020-04-16
山东化工(2019年18期)2019-10-16
石油与天然气化工(2019年1期)2019-03-06
科技资讯(2018年16期)2018-10-26
科技资讯(2017年12期)2017-06-09
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
