
QSAR modeling of benzoquinone derivatives as 5-lipoxygenase inhibitors
2019-05-26 03:35ShameeraAhamedVijishaRajanMuraleedharan
食品科学与人类健康(英文) 2019年1期
T.K.Shameera Ahamed,Vijisha K.Rajan,K.Muraleedharan
Department of Chemistry,University of Calicut,Malappuram 673635,India
Keywords:
ABSTRACT
1. Introduction
The biological properties of human 5-LOX have gained much attention from research because of its involvement in the pathogenesis of several diseases[1].The 5-LOX pathway is the source of potent pro-inflammatory mediators known as leukotrienes (LTs).5-LOX catalyzes the first two steps in leukotriene A4(LTA4)biosynthesis,in which the one is the addition of molecular oxygen into 1,4-cis–cis-pentadiene containing polyunsaturated fatty acids such as arachidonic acid and linoleic acid to give their hydroperoxy derivatives and the second step is the dehydration of this hydroperoxide to the key intermediate, short-lived epoxide leukotriene LTA4,which then converted to LTs[2].These LTs are essential mediators of excessive and chronic inflammatory and allergic disorders such as Rheumatoid arthritis, Gastroesophageal reflux disease,Atherosclerosis, Inflammatory bowel disease, and Autoimmune,ulcerative colitis,asthma,psoriasis and allergic rhinitis[3].Besides its important roles in inflammatory diseases,5-LOX is also involved in the development and progression of numerous types of cancer such as such as leukemia,pancreas,prostate,and colon cancer[4–8]. Therefore, the pharmacological intervention of the 5-LOX pathway to control the formation of LTs is a promising therapeutic strategy for LT-related diseases.
A wide variety of inhibitors have been reported as active against 5-LOX invitro studies.These compounds include phenols,aromatic compounds containing heteroatoms, carboxamides, hydroxamic acids, flavonoids, chalcones, etc. Based on the nature of action,these inhibitors have been classified into four types such as redox inhibitors, iron-chelator agents and non-redox or competitive inhibitors and 5-Lipoxygenase activating protein(FLAP)inhibitors[9,10]. Despite all this intensive effort, Zileuton is the only orally active drug that has been approved as 5-LOX inhibitor used for the treatment of asthma[11].However,zileuton itself has several side effects including liver toxicity and unfavorable pharmacokinetic profile [12]. So, the development of novel inhibitors having high inhibitory potency along with a favorable pharmacokinetic profile is the major challenge among the scientific community.
Drug discovery has been evolved from‘forward pharmacology’to rational drug design with the advancement of computational methods. Computer Aided Drug Discovery (CADD) revolutionize the area of drug design by identifying compounds with desirable characteristics, speed up the hit-to-lead process and improve the chances of getting your compound past the hurdles of preclinical testing. Now CADD is rapidly gaining in popularity, implementation and appreciation. Several publications have appeared in recent years documenting the rational design of 5-LOX inhibitors using computational methods such as ligand-based techniques like scaffold-hopping,pharmacological analysis,Quantitative structure-activity relationship(QSAR)and structure-based techniques like molecular docking,dynamic modeling[13].QSAR,a ligand-based CADD technique,is used to build computational or mathematical models which attempts to find a statistically significant correlation between various molecular properties of a set of molecules with their experimentally known biological activity.This method predicts the biological activity of known and unknown compounds by using statistical techniques and optimizing new lead molecules.
Several successful QSAR works were performed in recent years with the aim of formulating an excellent predictive model which composed of common chemical features among a considerable number of known 5-LOX inhibitors using conventional 2D QSAR methods such as Multiple Linear Regression(MLR),Principle component analysis(PCA)and partial least square regression(PLS).In literature, a few 3D-QSAR works for 5-LOX inhibitors also been documented [14,15]. Some of these studies [16,17] have used structure-based Comparative Molecular Field Analysis (CoMFA)and Comparative Molecular Similarity Indices Analysis(CoMSIA)as a tool to model the activity of human 5-LOX inhibitors. However,most of these studies have focused only on selective and nonredox type 5-LOX inhibitor and not on redox inhibitors.Non-redox inhibitors generally show diminished potency in a condition with elevated peroxide levels whereas redox inhibitors inhibit lipid peroxidation more effectively by scavenging peroxyl free radicals and suppressing the leukotriene formation[10].From the literature,it is observed that 5-LOX activity of redox inhibitors was enhanced by the presence of extended hydrophobic alkyl groups.Redox potency of these inhibitors might be directly linked with hydrophobicity.
In our previous study, we reported that the quantitative influence of extended hydrophobic alkyl groups of 3’, 4’-dihydroxyflavones derivative over the 5-LOX potency using CoMFA methodology [18]. In this work, we have tried to formulate QSAR models to investigate the interaction of a series of benzoquinone derivatives containing various lipophilic and bulky alkyl substituents reported by Rosanna Filosa et al.,[19]with the binding site of 5-LOX and predict their inhibitory activities. Here we reported four types of QSAR model: MLR based linear, RF and SVM based nonlinear 2D-QSAR models and CoMFA based 3D-QSAR model and the best two models were chosen to predict 5-LOX inhibitory activities. Furthermore, molecular docking studies have been carried out to rationalize the CoMFA model by demonstrating the common binding model of benzoquinone derivative with human 5-LOX model.
2. Materials and methods
2.1. Dataset
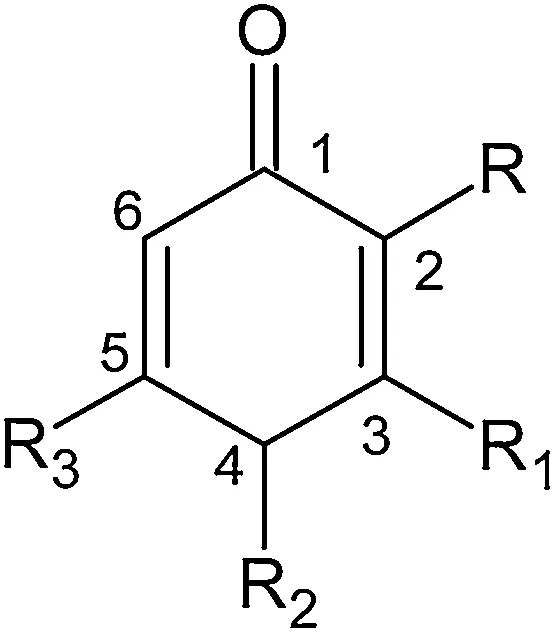
Fig.1. 2D chemical structure of benzoquinone core.
The dataset used in this study consisted of a series of benzoquinone derivative that has been reported as 5-LOX inhibitors in a cell-free assay using purified human recombinant 5-LOX enzyme by Rosanna Filosa et al.[19].2D structure of benzoquinone core is displayed in Fig.1.The experimental IC50values of all compounds in μM were converted into pIC50by taking-Log(1/IC50)and were used as the dependent variable.There were a total of 48 benzoquinone derivatives which are then split into a training set of 30 compounds for generating QSAR models and a test set of 11 compounds for validating the quality of the models.Remaining 7 compounds having IC50value greater than 10 μM were removed.The compounds in the test set were manually selected from the original pool of structures based on Y-response(dependent variable).This approach is based on the activity(Y-response)sampling.For maintaining uniform distribution, molecules with low, moderate and high activity were placed in both sets. Most active and least active molecules were retained in training set for better performance. All the structures and associated inhibitory activities are listed in Table 1.
2.2. Molecular modeling
All the 3D structures were drawn and built by Gauss View 05.Gas phase geometries were optimized using Density Functional Theory(DFT)[20]of three-parameter compound of Becke(B3LYP)[21,22]employing 6–31 G(d,p)basis set using Gaussian 09 program package[23].Harmonic energy calculations are carried out for confirming frequencies are all real. The lowest energy conformer of each compound was used for further analysis.
2.3. 2D-QSAR methodology
2.3.1. Calculation of 2D molecular descriptors
Descriptors are the mathematical representation of a molecule which contains different sources of chemical information transformed and coded to deal with chemical, biological and pharmacological problems.For the development of 2D QSAR models,various physicochemical descriptors are calculated for each of the compounds in the dataset by means of three different software such as e-DRAGON[24],PowerMV[25]and Gaussian 09.Different sets of 0D,1D,2D and 3D molecular descriptors are calculated with the help e-DRAGON software.Pharmacophore Fingerprint descriptors and Weighted Burden Number descriptors were computed by PowerMV software. Pharmacophore Fingerprint descriptors were built based on bioisosteric principles (Two atoms or groups that are expected to have roughly the same biological effect are called bioisosteres).Electronic and quantum chemical descriptors such as highest occupied molecular orbital(HOMO)energies,lowest unoccupied molecular orbital (LUMO) energies and molecular dipole moment were calculated by Gaussian 09.DFT based global reactivity descriptors provide the reactivity of chemical species regarding electronic features.Ionization potential(IP),electron affinity(EA),electronegativity(χ),electrophilicity(ω),softness(S),hardness(η)and chemical potential(μ)are the commonly used global reactivity descriptors.Koopman’s theorem approximates the negative value of HOMO and LUMO as ionization potential and electron affinity respectively. For the development of 3D-QSAR model steric and electrostatic descriptors were considered.The 2D-Descriptors used in the study given in Table 2.2D-QSAR models were generated using WEKA software[26].

Table 1 Structural formulae of compounds and their IC50 values.

Table 2 Descriptors used in the 2D-QSAR study.
2.3.2. Feature selection


Here, rcfis the average value of all feature-classification correlations,and rffis the average value of all feature-feature correlations.The Eq.(1)is,in fact,Pearson’s correlation where all variables have been standardized.
2.3.3. Multiple linear regression model
Multiple linear regression or MLR is a conventional and commonly used method in QSAR due to its simplicity, flexibility,reproducibility, and easy interpretability. The MLR attempts to model the relationship connecting a group of explanatory variables X and a response variable Y fitting a linear equation to observed data. The MLR model has a following mathematical form, given n observations:

Where Y is the pIC50of the benzoquinone derivatives, C0is the intercept and Cnare the regression coefficients of the descriptors Xn.Although MLR is computationally simple and the prediction models give a strong mechanistic interpretation,it is criticized for its lack of robustness in handling the non-linear data even though MLR models serve as the basis for some multivariate methods[28].
2.3.4. Support vector machines
Support vector machines(SVM)[29]are a bunch of supervised learning methods mostly used for classification and regression challenges. Classification of data using SVM involves by looking for a hyperplane in high dimensional space of independent variables that separate positive and negative data at an optimal distance using a non-linear kernel function. SVM methodology purely laid on maximizing the margin between a small subset of training instances(the support vectors)and the hyperplane.SVM methods are one of the most popular machine-learning methods in chemoinformatics.Several good reviews highlighting the application SVM in QSPR/QSAR studies particularly in drug design have been published[30]. The main advantages of SVM are: results are stable, reproducible, and largely independent of the optimization algorithm.The choice of a correctly configured kernel function is an important parameter to a successful SVM model.Polynomial kernel and Radical basis function(RBF)kernel are the two widely used kernels for solving classification problems. In this study, we employed a Polynomial kernel rather than RBF.WEKA Implements John Platt’s Sequential Minimal Optimisation (SMO) [31], an algorithm for training a support vector classifier.SMO normalizes all attributes;replace all missing values and transforms nominal attributes into binary ones.
2.3.5. Random forest
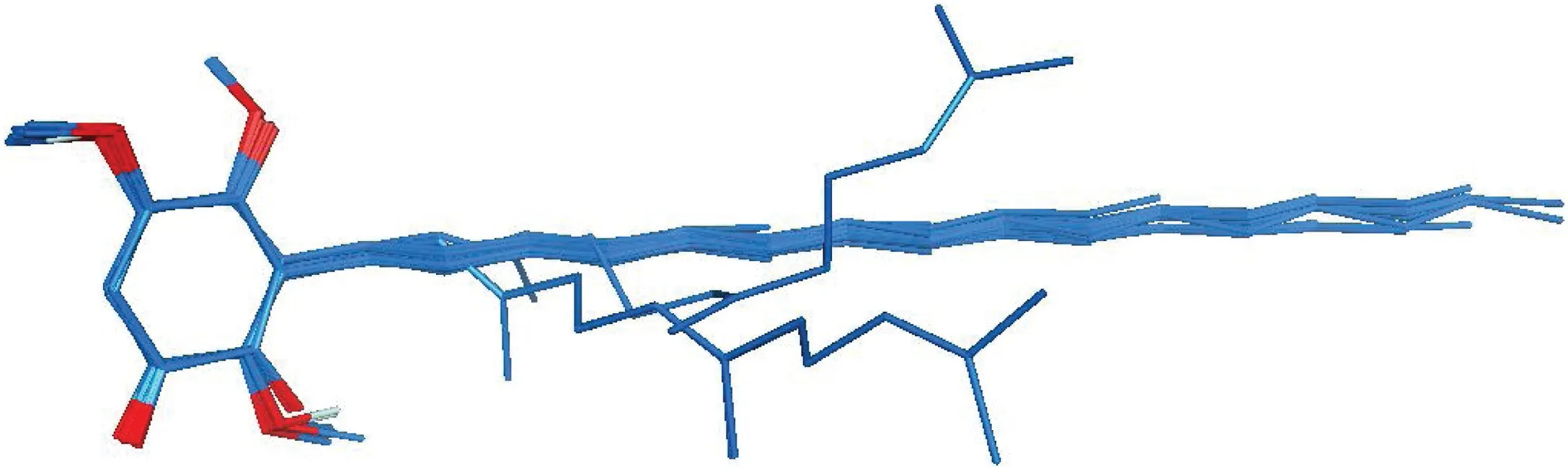
Fig.2. Alignment of 48 benzoquinone derivatives.
Random forest (RF) [32] (forest of decision trees) is an ensemble learning method of unpruned classification and regression trees such that each tree depends on the values of a random vector sampled independently and the same distribution of all trees in the forest.After a large number of trees is generated,each tree casts a unit vote for the most popular class. RF drawn sub-samples from original data with a replacement called as bootstrap sampling and fits trees to these samples. Here one-third of the data is left out of the bootstrap sample and used to testing while the rest form a training set. The prediction error internally estimates the performance of the developed model for the objects left out in the bootstrap procedure (out-of-bag estimation, OOB). Unlike Decision Tree which has relatively low prediction accuracy,RF displays some unique features that make it highly suitable for QSAR tasks.These include built-in estimation of prediction accuracy,the power of handling large dataset with higher dimensionality,measures of importance for each descriptor in the model, and a measure of similarity between molecules. This study explored the influence of the number of trees for the model’s predictive ability. Results showed that no significant change in the model’s predictive ability by increasing the number of trees beyond 1000.For all the descriptor set,the optimal tree values in the forest were set to 100.
2.4. Molecular modeling for 3D-QSAR
2.4.1. Molecular alignment and conformational analysis
Proper alignment of the compounds relative to one another is one of the most important steps in the 3D-QSAR analysis for obtaining valid molecular interaction field model. Energy minimized structures of molecules were aligned by the template-based method. The mixed alignment procedure in the combination of the Atom-based fashion and pharmacophore-based fashion was performed using Open3DALIGN software (version 2.27), an opensource tool capable of carrying out the multi-conformational,unsupervised rigid-body alignment of 3D molecular structures[33].The alignment procedure was executed by using all available molecules as possible templates. Hence, 48 alignments were produced, each obtained by superimposition on the corresponding template molecule.For each alignment,an O3 A score derived from the source code of the Open3DALIGN program is computed which indicates the quality of the superimposition. O3 A score for each alignment is given in supporting information S1. The alignment corresponding to the highest cumulative O3 A score was selected for further analysis.Fig.2 shows the best alignment in which compound 26a was selected as the template.
2.4.2. CoMFA analysis
CoMFA is a versatile method to describe 3DQSAR quantitatively.Open3DQSAR software (version 2.282) is open-source software available for high-throughput chemometric analysis of molecular interaction fields (MIFs) [34]. This study used Open3DQSAR to perform CoMFA analysis. The best alignment with compound‘26a’ as a template is placed in a 3D cubic lattice with 2 Å grid size and a 5.0 Å outgap.The steric fields were computed using sp3hybridized carbon atom probe with +1 charge. Similarly, electrostatic fields were computed using a volume-less probe.These steric and electrostatic interaction energies were considered as independent variables (CoMFA descriptors). Before creating of CoMFA model following pre-treatment operations were carried out to reduce the noise hidden in PLS matrix and hence reduced the computational time: 1) the minimum and maximum energy values of steric and electrostatic were set to a cutoffs value -30.0 and+30.0 kcal/mol, respectively. This pretreatment avoids infinity of energy values inside the molecule. 2) Low energy values(<0.05 kcal/mol) were set to zero in both fields. 3) Standard deviation set to <0.1 in order to improve the signal-to-noise ratio. 4)N-level variables that are variables which assume only N values across the training set were removed,most of which distributed on a small number of objects. This process avoids overweighting the importance of particular substituents present in a single molecule.Otherwise,it might negatively affect the whole model.5)The whole block of X or Y variables scaled by block unscaled weighting(BUW)technique.
Predictivity of CoMFA model can be significantly improved by appropriate variable clustering and selection procedures such as smart region definition(SRD)and fractional factorial design(FFD).These variable selection techniques selectively remove noisy variables with no predictability.The SRD procedure carries out variable grouping based on their closeness in 3D space in order to reduce the redundancy arising from the existence of multiple nearby descriptors which mainly encode the same kind of information [35]. FFD aims at selecting the variables which significantly increase the predictive ability (using the LOO, LTO or LMO paradigms), and can operate on both single variables or groups identified by a previous SRD run,thereby removing uninformative variables groups as performed in GOLPE[36].
PLS analysis implemented in Open3DQSAR was employed to obtain a correlation between the descriptors derived by CoMFA(independent variables) and pIC50values (dependent variable).Open3DQSAR generate a PLS model through the Nonlinear iterative partial least squares(NIPALS)algorithm[37].The statistical parameters like coefficient of determination (R2), Standard Deviation Error in Calculation(SDEC),Standard Deviation Error in Predictivity(SEDP) and F-ratio test were computed the overall significance of model(Eq.(3)–(5)).Moreover,the CoMFA color contour maps are derived for the steric and electrostatic fields.

Where,yobs,iis the experimental activity,ycalc,iis the estimated y in the calibration step and ypred,ipredicted the activity of the test set.The value corresponding to‘n’and‘p’is the number of samples in the training set and the number of components in the PLSR model respectively.
2.5. Statistical analysis and model validation
2.5.1. Internal validation
Three main cross-validations(CV)techniques such as leave-oneout (LOO), leave-two-out (LTO) and leave-many-out (LMO) were used to explore the reliability of statistical models.In LOO-CV,each time one compound is removed from the original training set,and a new model is built based on the rest of the set and this model is used to predict the activity of the omitted one.This procedure is repeated for whole compounds of data set. In LTO CV, two compounds are removed instead of one and the remaining procedure repeated as same as that of LOO. In the LMO method, each time 20% of compounds were removed randomly and the procedure was repeated 20 times and predicted their activities via the reduced model.Golbraikh and Tropsha reported that the LMO CV is much more robust than LOO-CV and also a high value of Q2is essential and important but not adequate for a predictive model [38]. The cross-validated R2,i.e.,Q2is given in Eq.(6).

The term PRESS is the sum of squared difference between experimentally observed activity and the activity predicted by a regression model estimated when the ithsample was left out from the training set and the SSY is the sum of squared differences between the experimental activity and the average experimental activity.According to Hawkins et al.,a valid statistical model should have high Q2value(Q2>0.5)and is evidence of the high predictive ability of the model[39].
2.5.2. External validation
The predictive power of the generated model was evaluated using an external test set of 13 molecules. The predictive correlation coefficient(R2pred)was determined according to the equation shown below,i.e.,Eq.(7).

SD is defined as the sum of the squares of the deviation between the experimentally observed activity of the test set compounds and the mean activity of the training set molecules.
2.6. Docking studies
Molecular docking methodology has been used to predict the best binding orientation of ligand molecule in the active site of receptor targets and recently it is the most used computational tool for drug designing and virtual screening.In this study,the crystal structure of human 5-LOX obtained from PDB database having PDB ID 3o8y was used for the docking study. AutoDock Vina [40]software was used to carry out molecular docking analysis. Optimized the dimension of the grid box to 20×20×25 Å with centre at-8.374,66.379,-1.009 for x,y and z respectively.AutoDock Vina uses a sophisticated gradient optimization in its local optimization procedure for rigid-flexible molecular docking.The AutoDock Vina represents the output of the docking result as Gibbs free energy of binding(ΔG).The top nine docking poses of each compound were visually analyzed and emphasized by ΔG values to rank the different conformations of the receptor-ligand complex. The ligand confirmation with lowest ΔG values was taken for further study.Protein-ligand complexes were visualized and analyzed using three different molecular modeling software LigPlus[41],Autodock tool 1.5.6[42],Chimera[43]and PyMol[44].

Table 3 Statistical data of optimal 2D-QSAR models.
3. Results and discussion
3.1. Feature selection and the 2D-QSAR prediction model
In order to develop accurate, robust and efficient 2D-QSAR models to predict 5-LOX inhibition activity of benzoquinone derivatives, a small subset of descriptors which represent the total set of descriptors has been identified through this work.We have employed the CFS optimization technique with the help CfsSubsetEval attribute evaluator of WEKA. Five descriptors show maximum correlation such as LUMO (lowest unoccupied molecular orbital- a quantum chemical descriptor), HBD06HBA(Pharmacophore fingerprints),WBNENH 0.75(Weighted Burden Number), X5A (average connectivity index of order 5) and JGI3(mean topological charge index of order 3-2D autocorrelation).Selected descriptors used in 2D-QSAR model with values were provided in supporting information S2.Based on these optimal features subset, the possibility of predicting 5-LOX inhibition activity was investigated with the help of MLR, SVM, and RF regression methods.The statistical performance of the optimum MLR,SVM and RF models using default parameters,is summarized in Table 3.
3.2. Statistical analysis of 2D-QSAR models
MLR Regression is the easiest and simplest technique to construct QSAR models but is also probably the least powerful. It can be used as a preliminary step before moving onto more complex algorithms.Linear regression works by calculating the coefficients for a line or hyperplane that best fits the training data.The best MLR equation obtained for the pIC50of the benzoquinone derivatives is based on the LUMO, HBD06HBA and JGI3 descriptors is given in Eq.(8).

Constructed MLR equations indicate the negative contribution of the above descriptors towards the prediction of 5-LOX activity of benzoquinone derivatives, i.e., 5-LOX receptor binding activity of these inhibitors might be decreased by increasing the LUMO energy value and HBD06HBA value as well as JGI3 value.The LUMO is the lowest energy level with an empty electron in the molecule. The energy of the LUMO is directly related to the electron affinity,i.e.,when a molecule undergoes a bonding interaction with protein; incoming electron pairs are received in its LUMO. Thus, the LUMO descriptor measures the electrophilicity of a molecule.Molecules with low LUMO energy are more able to accept electrons than those with high LUMO energy.The regression equations show that the low LUMO energy values positively influence the 5-LOX inhibition activity of benzoquinone derivatives.HBD06HBA is pharmacophoric descriptors account the hydrogen bond donor-acceptor features of benzoquinone that are thought to be responsible for pharmacological action.JGI3 is third order mean Galvez topological charge descriptor,the negative influence of this descriptor from this class to the ‘activity’suggested that a higher order charge index would not be beneficiary to the activity.
By analyzing the statistical parameter given in Table 3,we can say that the MLR model explains about 75%of the 5-LOX inhibitory activity variance of the training set. The predictive power of the MLR model was validated internally through cross-validation correlation coefficient Q2having 0.66 for LOO and 0.61 for LMO and externally through test set prediction using R2predvalue,0.54.The R2predvalue of the test set is less than 0.6; this indicates the MLR model is not satisfactory for prediction of the external test set.However, Stability of the QSAR models was analyzed by progressive Y-scrambling by taking the MLR model as a representative system. Y-scrambling (Y-randomisation) was applied to exclude the probability that our QSAR model performance could have occurred by chance.The Y-vector(pIC50)of the 30 compounds in the training set are sorted according to decreasing pIC50value, then these values were shuffled randomly and a new model was constructed.The shuffling within blocks is repeated 50 times. For each scrambling, an MLR and a CV model were computed. The obtained R2and Q2(LOO) values for random models should be less than that of obtained for the main initial model,then only we can accept the reliability of the main model. The results of the Y-scrambling test are given in supporting information S3. In all cases, the obtained random models have much lower prediction accuracies than the model based on the real data,indicating no apparent chance correlation in the QSAR model. The Average R, R2and Q2(LOO) for the 5-LOX inhibitors random model are around 0.41, 0.18 and -0.32 respectively.
SMO regression results were obtained using a popular algorithm-Sequential Minimization Optimization for regression(SMOreg). Statistical quality parameters were analyzed and it is found that Root means squared error(RMSE)generated by SMOreg were slightly higher than MLR model but this small difference may not be relevant and all other quality parameter values were slightly better than MLR model.R2predvalues of the test set are still less than 0.6;this indicates the SVM model is also not entirely satisfactory for prediction of the external test set. Hence it can be concluded that both MLR and SVM algorithms can be equally useful for internal prediction but not so good for external prediction.The experimental and predicted activities of the training set and the independent test set of MLR and SVM model are listed in Tables 5 and 6 respectively and corresponding scatter plot of observed vs. predicted values of pIC50of both the training and test set are shown in Fig.3A and B respectively.This data shows that the experimental and the predicted activities of inhibitors are not very close to each other.Most of the molecules show residual values greater than 0.4.
Then we made an RF regression model which works by constructing an ensemble of decision trees using training set and outputting mean prediction of the individual trees. One hundred trees and one seed were used for building the RF model for the study. Mean absolute error (MAE) measurement of the obtained model gives the magnitude of the error in prediction.For the training set,an MAE of 0.1154,RMSE of 0.1545 and R2,of 0.93 is obtained indicating that the better predictions. The cross-validated R2(Q2)is 0.52,and the R2predis 0.71,indicating good internal and external predictions of the developed RF model.The predicted pIC50values by RF model for training and test set are listed in Tables 5 and 6 respectively.Fig.3C demonstrates the correlation between experimental and predicted pIC50values by the RF model. These plots further reveal that the 2D-QSAR model based on the RF method is much better than that based on MLR and SVM methods. From all these results it can be concluded that the present RF model exhibits excellent predictive power from both the internal and external points of view concerning the prediction of the test sets.

Fig.3. Activity plots of observed vs.predicted pIC50 of training and test set of 5-LOX inhibitors by the A)MLR,B)SVR,C)RF and D)CoMFA models.
3.3. Statistical analysis of CoMFA models
Using the training set of 30 benzoquinone derivatives, CoMFA model with five PLS components was built and then, the external test set including 11 compounds was used to evaluate the reliability and applicability of the built model. Statistical quality parameters associated with CoMFA models based on FFD procedures for noise reduction in the input data is listed in Table 4. The analysis of these parameters revealed that the best CoMFA model was obtained with a combination of steric and electrostatic fields.Thepercentage contribution of the steric field and electrostatic field to the PLS model is 77 and 23%respectively.i.e.,more than 75%contribution was observed from the steric field, indicating that steric interaction is essential to the binding of benzoquinone analogs with 5-LOX. CoMFA model gave good cross-validated correlation coefficient (Q2) for LOO, LTO, and LMO as 0.5976, 0.5851 and 0.5361 respectively, indicating an excellent internal predictive power of the established model. The non-cross-validated PLS analysis with the five components resulted in a conventional R2of 0.8489, F of 26.97 and SDEP of 0.2203 for CoMFA model was found to reasonable.The values of experimental and predicted activities along with the residual values of the training set and test set molecules are summarized in Tables 5 and 6 respectively. The scatter plot of observed vs. predicted values of pIC50of both the training and test set of CoMFA models is shown in Fig.3D. This data shows that the experimental and the predicted activities of inhibitors are very close to each other.Most of the molecules show residual values less than 0.4.This graphical representation again conforms the good predictive power of the established model and also indicated that the developed CoMFA model is reliable,and could be used in designing new inhibitors.
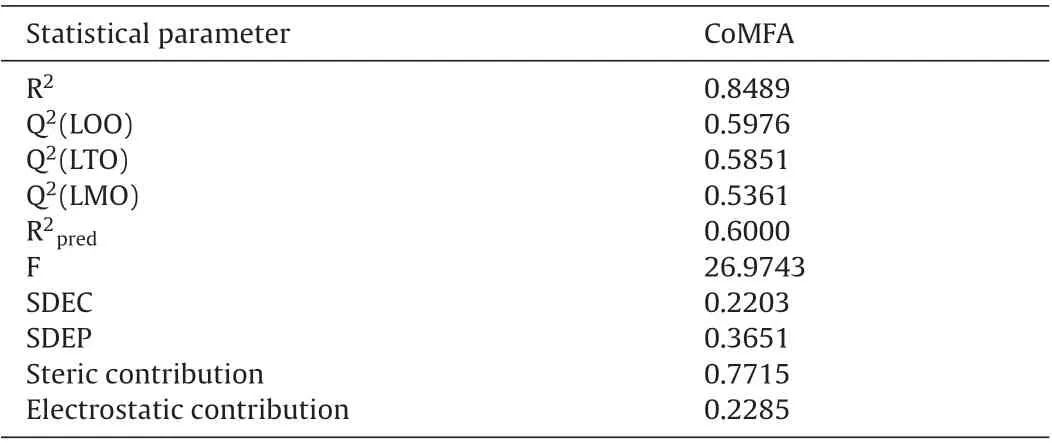
Table 4 Statistical data of optimal CoMFA model.
3.4. Graphical interpretation of the CoMFA contour maps
The most significant advantage of CoMFA is that it generates 3D contour plots around the molecules.These contour maps are used to identify regions in MIFs of the molecules included in the trainingset where any change in the steric and electrostatic field might affect the biological activity and they also provide hints for the modification required to design new molecules with better activity.The CoMFA steric and electrostatic contour maps are shown in Fig.4. The Green and yellow contours represent the steric fields.In detail, the green region in the steric contour maps indicates an area where the bulky groups are favored for activity while the yellow contours represent regions where the bulky groups are not favored for the activity. The red and blue contour represent electrostatic contour maps.The blue contour defines a region of space where positively charged substituent increases activity, whereas the red contour defines a region of space where negatively charged substituent increases activity.

Table 5 The experimental and predicted pIC50 values of the training set.
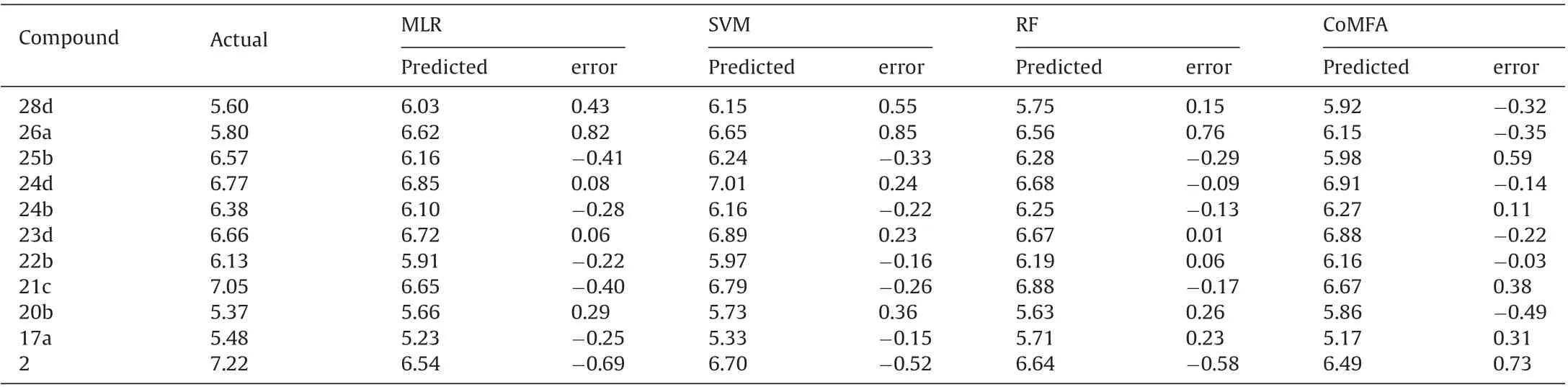
Table 6 The experimental and predicted pIC50 values of the test set.

Fig.4. PLS contours obtained from 3D-QSAR model of 5-LOX inhibitors.(A)CoMFA steric contour maps(B)CoMFA electrostatic contour maps.
These contour maps give us some general insight into the nature of the receptor-ligand binding region.Three green plots were found around the middle of the ‘n-alkyl’ residue in position 3 indicate large groups in this region(C10-,C11-,C12-,C13-,C14-,C15-and C16-) is favorable to increase the activity of the ligand. To justify this,we could say that the compounds 20d,22d,24d,and 26d with C10-, C12-, C14-, and C16-n-alkyl chains, respectively inhibited 5-LOX with low IC50values between 0.17 and 0.19 μM than those of the compounds 17d, 18d and 19d with C4-, C6- and C8-alkyl substituent respectively.The long,bulky alkyl group in these regions are significant for a potentially active ligand. This observation is in agreement with general findings of parent literature showing that the potency of(poly)phenol-based 5-LOX inhibitors is often enhanced due to increasing the lipophilicity.This sterically crowded alkyl group may bring a hydrophobic nature to the parent benzoquinone thereby enhances the activity.The small yellow contours at the tail portions of the alkyl residue in position 3 show that too long alkyl chains (greater than C16-) might have a negative influence on its activity. A large yellow contour at the 5thposition indicates that the OH group is preferable at this position as compared to OCH3.This result is confirmed by the lower activity of compounds in‘b series’like 18b,26b,and 27b(Table 1)which are the derivative of benzoquinone methylated the hydroxyl in 5thposition.



Fig.5. 2D view of the binding interaction of Compound 2(Embelin)with 5-LOX.
3.5. Molecular docking analysis
Interaction of benzoquinone derivative with 5-LOX was observed to get the view of ligand conformational change when undergoes docking.Since a co-crystal ligand was absent for 5-LOX,Prediction of the size and spatial orientation of the ligand binding sites of proteins was a major challenge. The active site of 5-LOX crystal structure was reported in literature characterized by an elongated cavity which is surrounded by 5-LOX specific amino acids Tyr 181,Ala 603,Ala 606,His 600 and Try 364 and all LOX conserved residues like Leu-368,373,414,607 and Ile-406[45].We also have confirmed the active site by identifying the possible cavity of 5-LOX with the help of Pocket-Cavity Search Application POCASA and optimized the dimension of the grid box[46].
Using the optimized grid box and through molecular docking process,the interaction between protein 5-LOX and benzoquinone derivatives were deduced in the form of binding affinity value given in Supporting information S4.The binding mode between the natural derivative of benzoquinone‘Embelin’(compound 2)and 5-LOX(Fig.5)reveals that inhibitory mechanism of compounds is almost similar to the typical inhibitory mechanism of a good inhibitor for 5-LOX, which should have a polar head and a hydrophobic body.The polar OH-and C O groups at the benzoquinone head portion interact with the polar amino acids of 5-LOX by forming hydrogen bonds with His 600,Gln 363 and Leu 420 residues and can be seen in green dotted line. This observation is compatible with CoMFA electrostatic contours found around benzoquinone head portion indicating these regions are favorable may be due to its interaction with polar amino acids of the target protein. This compound also forms hydrophobic interactions with the protein through its nonpolar long alkyl part with residues like Leu 368, Ile 415, Phe 421,Phe 359, Leu 414, Leu 607, Phe 177. These findings again support the CoMFA result which has shown the importance of long,bulky alkyl group at the position 3 for a potentially active ligand.
4. Conclusions

Conflict of interest
No conflict of interest to declare
Acknowledgments
The authors T.K.Shameera Ahamed and Vijisha K.Rajan express their sincere gratitude to Human Resource Development Group Council of Scientific&Industrial Research(CSIR),India and University Grants Commission(UGC),India,respectively for the financial support. The authors are thankful to the Central Sophisticated Instrumentation Facility (CSIF) of the University of Calicut for the Gaussian 09 software support.
Appendix A. Supplementary data
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.fshw.2019.02.001.
- 食品科学与人类健康(英文)的其它文章
- Microalgae:A potential alternative to health supplementation for humans
- Autophagy-associated signal pathways of functional foods for chronic diseases
- Screening of potential GCMS derived antimigraine compound from the leaves of Abrus precatorius Linn to target“calcitonin gene related peptide”receptor using in silico analysis
- Rapid and easy determination of morphine in chafing dish condiments with colloidal gold labeling based lateral flow strips
- Optimization of process conditions for drying of catfish(Clarias gariepinus)using Response Surface Methodology(RSM)
- High uric acid model in Caenorhabditis elegans