
Autophagy-associated signal pathways of functional foods for chronic diseases
2019-05-26 03:35JinfengXieJilingLingNingChen
食品科学与人类健康(英文) 2019年1期
Jinfeng Xie,Jiling Ling,Ning Chen
a Graduate School,Wuhan Sports University,Wuhan 430079,China
b Tianjiu Research and Development Center for Exercise Nutrition and Foods,Hubei Key Laboratory of Exercise Training and Monitoring,College of Health Science,Wuhan Sports University,Wuhan 430079,China
Keywords:
ABSTRACT
1. Introduction
With the development of the times, inappropriate diet and lifestyle lead to a high incidence of chronic diseases,which seriously threatens people’s health and life quality [1]. Currently, the most common and high-frequent chronic diseases in aging population include diabetes, obesity, metabolic cardiovascular diseases, and neurodegenerative diseases. A large number of experiments have demonstrated that functional foods associated with autophagy signal pathways can execute health promotion, and prevention and treatment of chronic diseases, but the underlying molecular mechanisms are unclear [2–4]. Many scholars are exploring the underlying molecular mechanisms of these bioactive components for functional foods, elucidating autophagy-related signal pathways of functional foods, and screening the novel and effective targets of functional foods for health promotion and prevention and treatment of these chronic diseases[2,5–7].
2. Functional foods
Currently,the demand for health concerns by people is increasing all over the world. The mounting consciousness is associated with the vital role of functional foods in health promotion,and the prevention and treatment of chronic diseases.Thus,an increasing number of people have the high requirement for healthy foods or functional foods in many parts of the world[8].
Chinese medicinal foods have been favorite for thousands of years. Nowadays, the development in scientific research sustains the idea that the diet not only provides necessary nutritional support, but exerts a beneficial role in some chronic or metabolic diseases[9,10].Initially,the concept of functional foods is to seek foods with the function of treating diseases [11]. From 1984 to now, functional food has changed its meaning due to different culture backgrounds, and confused with nourishment sometimes[12]. In 2014, in the 17th international conference of functional food in health and diseases, functional food has been provided a new definition as natural or processed foods containing known or unknown bioactive components at the effective non-toxic dosage to execute clinically-proven or documented health benefit for the prevention,management or treatment of chronic diseases beyond the basic nourishments[11].Therefore,the health beneficial effects and proper dosages of bioactive components in functional foods and the management of chronic diseases by using functional foods have gained tremendous attraction and systematic investigation.Bioactive compounds are usually regarded as the most significant ingredients in functional foods,which are not only necessary,but also indispensable for health promotion, and the prevention or treatment of chronic diseases. With the development of chemical and biological technology,specific bioactive compounds have been isolated and purified, and the relationships between specific bioactive compounds and chronic diseases have been elucidated, as well as a growing consciousness associated with the importance of specific bioactive compounds in functional foods is enhanced due to their pleiotropic effects including antioxidant,anti-inflammatory, immune-enhancing, hypolipidemic, glycemicregulating, cytoprotective, and neuroprotective functions [5].Although the shortcoming of functional foods on the prevention and treatment of chronic diseases may be present only after longterm or sustained consumption, functional food as a preventive and therapeutic natural or processed food is not a substitute but a coordinator for our daily diet.
3. Autophagy
Autophagy as an evolutionary-conserved process is responsible for lysosomal degradation and continuous removal of mis-folded toxic protein aggregates and damaged or dysfunctional cell organelles,thus executing its cytoprotective role[13,14].Growing evidence has demonstrated that autophagic capacity to degrade harmful aggregated proteins in cells presents a gradual reduction as the extension of age or aging process [15]. Furthermore, dysfunctional autophagy has also been linked to a series of chronic diseases or aging-associated diseases [16–19].Autophagy is one of two major known routes for the clearance of aberrant components in eukaryotic cells. As a primary proteolytic system, mis-folded proteins and damaged cellular organelles are sequestered within double-membrane vesicles to form autophagosomes, and sequentially bind with lysosomes to form autolysosomes for accomplishing the degradation of these damaged or dysfunctional cellular contents and the recycling.This highly-conserved autophagic process plays a vital role in the adaptation of cellular stress and the maintenance of cellular homeostasis[20,21].More than 30 autophagy-regulated genes(Atgs)have been screened and identified in yeast and humans for controlling the whole autophagic process[22,23].
There are three major pathways for the regulation of autophagy.The first one is modulated by nutrients or nutrient-induced insulin,which is dependent on mammalian target of rapamycin (mTOR).Class I phosphatidylinositol 3-kinase (PI3K) can activate protein kinase B (Akt) and mTOR. As a critical negative regulatory factor of autophagy, mTOR can inhibit ULK1/2-mAtg13-FIP200(yeast homolog Atg1-Atg13-Atg17)complex[24],thereby recruiting other related proteins for the formation of autophagosomes.The second one is regulated by Beclin1 (yeast homolog Atg6),which can combine with class III PI3K Vps34 (vacuolar protein sorting 34)to form Beclin1-Vps34 complex.The phagophore lipid membrane needs Beclin1-Vps34 complex activated by ULK1/2-mAtg13-FIP200 complex for its elongation[25].Beclin1,a regulator involved in both autophagy and apoptosis,can bind with apoptotic inhibitors such as B-cell lymphoma/leukemia-2(Bcl-2)and Bcl-XL to inhibit autophagy under the condition of adequate nutrition[26].The third one is controlled by two ubiquitin-like proteins, Atg12 and LC3 (microtubule-associated protein on light chain 3, yeast homolog Atg8) [25], for controling the elongation of phagophore membrane and the formation of autophagosomes.On the one hand,Atg12, activated by Atg7 and transferred to Atg10, can conjugate with Atg5 to form Atg12-Atg5 complex for the further association with Atg16.Finally,Atg12-Atg5-Atg16 complex is attached to the phagophore and separated when autophagosomes are entirely formed.On the another hand,LC3,cleaved by Atg4,is regulated by Atg7 and Atg3 to become LC3-II[27].LC3-II is connected with lipid phosphatidylethanolamine(PE)to promote the elongation and closure of the membrane of autophagosomes(Fig.1).
In the basal level, autophagy selectively targets dysfunctional intracellular components, such as mitochondria, endoplasmic reticulum, peroxisomes, and so on [28,29]. Although autophagy as a cellular protective mechanism can eliminate dysfunctional components, the abnormal functional status of autophagy such as overactive autophagy, insufficient autophagy, or impaired autophagic flux could result in cell dysfunction[15].What’s more,autophagy can be out of control to maintain cellular functions in numerous chronic diseases including diabetes,obesity,cardiovascular metabolic disease,and neurodegenerative diseases[30,31].
4. The regulatory roles of autophagy in chronic diseases
Many chronic diseases are correlated with the disrupted metabolic processes including metabolic disorders or active metabolism, which is involved in autophagy as a vital regulatory factor for three primary energy substances such as glucose, lipid and protein [32,33]. A common pathophysiological alteration in chronic diseases is the accumulation of harmful contents such as reactive oxygen species(ROS),damaged organelles,protein aggregates,lipid droplets,and senescent cells.In contrast,the enhanced autophagic flux or improved functional status of autophagy may be beneficial to degrade harmful substances for the alleviation of chronic diseases.
4.1. Diabetes and autophagy
Diabetes is characterized by hyperglycemia resulting from insulin deficiency, insulin resistance, or both, which is involved in the regulation of autophagy for maintaining β-cell function and survival, and sufficient insulin [34,35], as also evidenced by tissue-specific Atg knockout mice displaying morphological abnormality and degeneration of islets, and accumulated protein p62/sequestosome1(p62/SQSTM1)and polyubiquitinated protein in β-cells, as well as impaired insulin secretion [36]. Beta cells are highly susceptible to hyperglycemia-induced oxidative stress,thereby resulting in reduced insulin secretion and accelerated βcell death[37].In addition,nuclear factor erythroid 2-related factor 2 (Nrf2) can protect β-cells from damage and apoptosis through regulating the transcription of autophagy genes,and the knockout of Nrf2 results in an obvious decrease in the number of β-cells[38].Under the condition with oxidative stress,autophagy can execute the selective degradation of dysfunctional mitochondria, termed as mitophagy. As us well known, long-term exposure to glucose and fatty acids, can lead to glucolipotoxicity and ROS production,thereby resulting in the accumulation of dysfunctional mitochondria [39]. Thus, the management of glucolipotoxicity, oxidative stresses,and dysfunctional mitochondria by mitophagy is essential for the protection of β-cells.
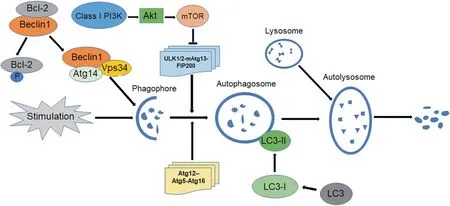
Fig.1. Major regulatory processes of autophagy,including that mTOR signal pathway inhibits the formation of autophagosomes;Beclin1-Vps34-Atg14 complex stimulates the elongation of lipid membrane phagophores;lysosome binding contributes to the formation of autolysosomes for the degradation of dysfunctional cellular contents.
Except for β-cell dysfunction, insulin resistance in a wide range of tissues is a hallmark of type II diabetes mellitus (T2DM).Induced autophagy acts as an important protector against oxidative stress in insulin-target tissues such as liver, adipose tissue and skeletal muscle [40]. In addition, genetic Keap1 knockdown or pharmaceutical-induced Nrf2 could improve insulin sensitivity in diabetes and obesity, thereby suggesting Nrf2/Keap1/ARE signal pathway as a therapeutic target in T2DM [41]. Similarly,Atg7-deficient mice show reduced β-cell number, impaired glucose tolerance, and declined insulin secretion [42], suggesting autophagy as a crucial target in treating diabetes. Under diabetic conditions, p62 and lysosomal protein Lamp2a levels usually are higher than non-diabetic controls, indicating the reduced or dysfunctional autophagic flux.In endothelial cells of diabetic models,endothelial nitric oxide synthase (eNOS) is down-regulated; but the beneficial effect of spermidine on the activation of eNOS can be blocked by autophagy inhibitors such as bafilomycin or 3-methyladenine, indicating that inadequate autophagy can lead to endothelial dysfunction in diabetes [43]. Based on above discussion,the treatment of diabetes may have high requirements for the induced autophagy.
4.2. Obesity and autophagy
Obesity patients in mounting number have been recognized as a global health concern, with more than one-third of overweight or obese people worldwide[44].The obesity is correlated with the accumulated lipid droplets,protein aggregates,and damaged mitochondria as the preferred substrates of autophagy. Among obese patients, excessive nutrition can suppress autophagy by inhibiting adenosine 5'-monophosphate (AMP)-activated protein kinase(AMPK) and mTORC1 [45]. No matter genetic and dietary models of obesity present the suppressed autophagy, especially the reduced Atg7 in liver[46].Meanwhile,the increased p62 and LC3-II in liver compromised autophagic flux in primary hepatocytes are also detected.Therefore,the blockage of autophagosome maturation may be the pathogenesis of obesity. In addition, autophagy deficiency in hypothalamus could impair the central control of energy balance[47].Therefore,enhanced autophagy may serve as a compensatory mechanism for limiting excessive inflammation and adipogenesis[48,49].
Similarly, obesity-induced lipotoxicity can down-regulate AMPK signaling, thus decreasing the formation of autophagosomes in macrophages and hepatocytes[50,51].ER stress provoked by obesity and lipotoxicity can induce autophagy via multiple mechanisms. Other stresses associated with obesity, including inflammation and oxidative stress, can also activate autophagy[9,52]. Autophagy induction in this context can be viewed as the partial cellular defense mechanism for maintaining cellular homeostasis under obesity-associated stresses.
4.3. Metabolic cardiovascular disease and autophagy
Autophagy,as essential for maintaining cardiovascular function,can be induced in metabolic cardiovascular diseases such as cardiac hypertrophy, heart failure, or ischemic heart disease [53]. However,the decreased autophagic flux is observed in glycogen storage disease-related cardiomyopathy.Ischemia/reperfusion(I/R)injury after myocardial infarction is characterized by the accumulation of free radicals, mitochondrial damage, and abnormal cardiovascular function. During cardiac ischemia phase, AMPK can activate autophagy through up-regulating AMPK-mTORC1-ULK1 signaling or directly phosphorylating and activating ULK1,thereby enabling the initiation of autophagy [45,54]. Free fatty acids and amino acids are released via the degradation process of autophagy, for which is essential for the survival of cardiomyocytes. During cardiac reperfusion phase,excessive autophagy may be detrimental to the survival of cardiomyocytes. At cardiac reperfusion phase, the rescuing of injured tissues can result in the consequence either cell preservation (preconditioning) or cell death (I/R injury). In simulated I/R(sI/R)cardiomyocyte model,autophagosome formation and lysosomal degradation are impaired.Conversely,increased autophagic level of cardiomyocytes can execute the protection of sI/R injury.Up-regulated Beclin1 will improve autophagic flux and reduce pro-apoptotic Bax activation,but RNA interference(RNAi)of Beclin1 can promote Bax activation and apoptosis[55].Autophagic death of cardiomyocytes is evidenced by the observation that Beclin1 siRNA reduces cell death in cardiomyocyte I/R injury[56].
The enlarged volume of cardiomyocytes, disarranged sarcomeric structure and enhanced protein synthesis in pathological cardiac hypertrophy are the responses to various biomechanical and physiological or pathological stimuli. However, long-term exposure to these stimuli can accelerate the progression of heart failure.Accumulating evidence has revealed the possibility of regulating autophagy as a therapeutic goal of cardiac hypertrophy.The induced autophagy by inhibiting mTOR and activating AMPK can prevent cardiac hypertrophy and improve cardiovascular function through pharmacologic treatments [57,58]. Overexpressed miR-99a activates autophagy via inhibiting mTOR signaling, and improves cardiomyocyte function and survival after myocardial infarction [59]. But, miR-132, an autophagy-inhibiting miRNA,rescues stress overload-induced cardiac hypertrophy and heart failure in mouse model [60]. The miRNAs with regultory functions of autophagy may be also the novel and effective therapeutic targets for stress-triggered cardiac hypertrophy. In apolipoprotein E (ApoE)-null mice, specific knockout of Atg7 or Atg5 genes for resultant defective autophagy could accelerate atherosclerotic plaque formation, and stimulate inflammatory reaction,interleukin-1β production, plaque cell death, and fibrous cap thinning [61,62]. Similarly, the accumulation of p62/SQSTM1 in Atg5-null macrophages is also observed. According to mouse and human atherosclerotic samples, p62/SQSTM1 and polyubiquitinated proteins display an increased level[63].Above all,defective autophagy may be a contributing factor in the progression of atherosclerosis.
4.4. Neurodegenerative diseases and autophagy
In neurons, aggregated proteins can lead to synaptic impairment, cellular organelle damage, and neuronal cell death. The neurodegenerative diseases including Alzheimer’s disease (AD),Huntington’s disease (HD) and Parkinson’s disease (PD) are observed to have considerable accumulation of intracytoplasmic autophagosomes and aggregated proteins in neurons[64,65],suggesting that autophagic machinery cannot eliminate aggregates in a coordinated manner due to the impaired autophagic flux [66].Although these disorders reveal different mutated proteins,protein aggregation and its underlying mechanism are similar.In general,inducing autophagy may result in the alleviation of pathological symptoms[67–69].
In animal models with AD, the down-regulated Beclin1 and other core autophagic genes could result in the accumulation of amyloid beta (Aβ) [70–72]. Nuclear receptor binding factor 2(NRBF2), a significant member of PI3K complex with positively regulatory function for autophagy, could be significantly downregulated[73].mTOR signaling is also involved in AD pathogenesis.Increased mTOR activity can lead to deficient autophagy, thereby inducing the overexpression and abnormal phosphorylation of Tau[74].Similarly,mTOR-independent autophagy inducers have been applied to reduce cerebral amyloid plaque burden and Aβ level[75,76], and enhance the clearance of amyloid precursor protein(APP) C-terminal fragments through activating Atg5-dependent autophagy[77].
HD as an inherited disorder is due to the accumulation of misfolded huntingtin (HTT) and the expansion of the polyglutamine(poly Q) tract in N-terminus of HTT [67]. Increased Beclin1 in HD mouse models can promote neurodegenerative effects[78].Meanwhile,mTOR inhibitors also can reduce the aggregation of mutated HTT protein and cell death by inducing autophagy[79].
PD is charactered by the accumulation of Lewy bodies (LBs)consisting of aggregated α-synuclein (α-syn) that can lead to the impaired axonal transport [80,81]. Previous findings have documented the blockaded autophagy in PD and the induced autophagy for executing neuroprotective function via AMPK/SIRT1 signal pathway; therefore, the overexpression of Beclin1 in PD mouse models can rescue the neurodegeneration[82](Fig.2).
5. Autophagy-associated functional foods in chronic diseases
The regulation of autophagy may prevent the occurrence,delay the progression, and decrease the severity of certain chronic diseases. It will provide the fundamental rationale for the research and development of autophagy-associated functional foods.As us well known,bioactive compounds in functional foods are the primary ingredients. Accumulating evidence have displayed that an increasing number of autophagy-associated natural products play a crucial role in the prevention and treatment of chronic diseases through modulating oxidative stress response,inhibiting apoptosis,and regulating autophagy functional status[4,83].As follows,there are listing several commonly investigated autophagy-associated bioactive components in functional foods:
5.1. Resveratrol
Resveratrol (RSV) usually exists in berries, nuts, grapes, and other plants [84] as a kind of polyphenol with pleiotropic action including antioxidant activity, anti-inflammatory role, cytoprotective effect and neuroprotective properties [2,85,86], thereby executing its beneficial functions in these common and highfrequent chronic diseases. Cell experiment has proved that RSV could induce autophagy based on the observation with the increased number of LC3-II puncta, the up-regulation of Bcelin1,and the enhanced LC3-II/LC3-I ratio[87].In contrast,chloroquine,an autophagy inhibitor,could reduce these effects of RSV.In addition,RSV can significantly induce autophagy by activating sirtuin3(Sirt3) protein and its downstream enzyme for responding to ER stress.On the other hand,Sirt3-siRNA transfection could partially inhibit autophagy, thus confirming the autophagy-inducing function of RSV [87]. Similarly, RSV can activate Sirt1 and AMPK to rescue pancreatic β-cell dysfunction and improve insulin sensitivity[6,88].
At present, RSV also serves as a natural compound exerting numerous positive effects on diabetes,and is widely investigated in animal models and diabetic population.RSV can activate autophagy and attenuate apoptosis to protects cardiac cells of diabetic models through regulating AMPK/mTORC1/p70S6K1/4EBP1 and JNK signal pathways for dissociating Beclin1-Bcl-2 complex [89]. In obesity model, RSV has been shown to suppress inflammatory reaction, improve insulin sensitivity, and decrease lipid deposition, thereby inhibiting the oxidation of low-density lipoprotein(LDL) and the aggregation of platelets [90]. What’s more, RSV can activate autophagy by regulating AMPK-mTOR signal pathway to significantly reduce palmitic acid (PA)-induced endothelial ROS level in human aortic endothelial cells [91]. Similarly, RSV has also been reported to attenuate the symptoms of AD by inhibiting Aβ generation and aggregation, promoting Aβ clearance, and modulating Tau-associated neuropathological processes including inhibiting abnormal Tau phosphorylation and Tau aggregation[92,93]. RSV displays the protective effect against AD by activating autophagy through regulating AMPK/Sirt1 or mTOR-dependent signal pathway [2]. The induced autophagy will beneficial for the clearance of Aβ and lipopolysaccharide (LPS), thereby suppressing inflammatory cytokines including interleukin 1 beta (IL-1β),tumor necrosis factor alpha (TNF-α) and nuclear factor kappa-B(NF-κB).Autophagy is also partially regulated by activating tyrosyl transfer-RNA (tRNA) synthetase (TyrRS)-poly (ADP-ribose) polymerase 1 (Parr1)-Sirt1 signal pathway to alleviate neurotoxicity caused by β-amyloid protein fragment 25–35(Aβ25–35)[94].During PD treatment,RSV can induce autophagy through regulating the AMPK-Sirt1 signal pathway to accomplish neuroprotective effects in a rodent model of PD[95].Dopamine toxicity for cells expressing mutant HTT could be protected in the presence of RSV, which may be correlated with the rescued formation of Atg4-mediated autophagosomes[96].
5.2. Epigallocatechin-3-gallate
Epigallocatechin-3-gallate (EGCG) has been gained increasing attention due to its antioxidant capacity,and its capability to regulate insulin sensitivity,lipid metabolism,inflammatory response,vascular endothelial function, and neuroprotective roles [97–99].As a bioactive polyphenol in green tea, EGCG can alleviate the progression of metabolic diseases through activating autophagy,thereby reducing lipid accumulation in vascular endothelial cells[100].EGCG also can induce autophagy to suppress the accumulation of lipid droplets induced by palmitate [100]. Further studies have shown that EGCG can induce autophagy with the typical up-regulation of autophagic biomarkers such as LC3-II, Atg7, and Beclin1. Compared with the control in adipocytes, the enhanced phosphorylation of AMPK indicating an energy-depleted state can be observed during EGCG treatment, thereby significantly reducing triglycerol by 25%,mitochondrial membrane potential by 56.8%and intracellular ATP level by 49.1%[101],which supports the therapeutic potential of EGCG for obesity and obesity-related metabolic diseases [101]. In spontaneously hypertensive rats (SHR), EGCG can significantly alleviate I/R injury via PI3K-nitric oxide(NO)signal pathway to reduce infarct size and improve cardiac function[102]. ECGC also can activate Nrf2 to suppress oxidative stress,inflammation, and cell death [103]. It has been well documented that EGCG alleviates myocardial I/R injury and the dysfunction of post-ischemic myocardial cells in vitro and in vivo[104,105].EGCG also can execute neuroprotective effect by activating autophagy as evidenced by increasing LC3-II and blocking p62 as well as inhibiting Bax and cytochrome C(Cyc)during neurodegenerative process[106]. Long-term treatment with EGCG can reduce amyloid beta 1–42(Aβ1–42)level through inducing autophagy,which is also validated by the reversed observation in the presence of chloroquine as an inhibitor of autophagic flux[106].
5.3. Curcumin
Curcumin, the primary therapeutic component of turmeric,has been known for its anti-inflammatory and antioxidant activity [107]. It also has hypolipidemic, cardiovascular protective,and neuroprotective effects [108–110]. Pre-treatment with curcumin can significantly down-regulate the expression of NF-κB and ROS,and increase superoxide dismutase,catalase,and glutathione.Meanwhile,curcumin pretreatment also can defend mitochondrial dysfunction, activate caspases and apoptosis in cells [107]. When rats with diabetic cardiomyopathy are orally administrated with curcumin,the fibrosis,oxidative stress,inflammation and cell death can be obviously attenuated;moreover,the decline of Akt and GSK-3β can be improved by curcumin [111]. Curcumin treatment can significantly suppress the level of I/Rinduced apoptosis and activate autophagy by inducing the expression of Bcl2 and Bax and inhibiting the expression of Beclin1 and Sirt1, respectively, which will promote cell survival as confirmed in H9c2 myocytes [112]. Curcumin significantly increases not only the mRNA levels of mTOR,LC3 and Beclin1,but also the protein expression of p-mTOR,LC3-II,and Beclin1,as well as LC3-II/LC3-I ratio.In model rats with cardiac hypertrophy and fibrosis,curcumin treatment attenuates the interstitial fibrosis of heart through mTOR-mediated autophagy signal pathway [113]. Furthermore, autophagy induced by AMPK-mTOR signaling has the potential of preventing I/R-induced damage of cardiomyocytes[114].
Curcumin can enhance autophagy to protect human endothelial cells from injury caused by oxidative stress.Pretreatment with curcumin can up-regulate LC3-II and Beclin1,promote the formation of autophagosomes,and accelerate the degradation of p62 for improving cell survival under the circumstance of oxidative stress.Curcumin also can induce autophagy in human umbilical vein endothelial cells(HUVECs),via not only inhibiting PI3k/Akt/mTOR signal pathway,but also elevating the level of cytoplasmic acetylation of forkhead box protein O1(FoxO1)as well[109].In contrast,when FoxO1 is knocked by shRNA, the protective effect and the autophagic process in the presence of curcumin could be inhibited. Curcumin may protect cells against oxidative stress through inducing autophagy via Akt/mTOR signal pathway[115].Curcumin can significantly induce autophagy via down-regulating mTOR signaling to decrease the accumulation of α-synuclein in A53 T cells[116].According to previous reports,curcumin not only can attenuate cognitive disorder, but also can inhibit the generation of Aβ and induce autophagy by down-regulating PI3K/Akt/mTOR signal pathway to accomplish neuroprotective effect in AD mice(APP/PS1 double transgenic mice) [117]. Therefore, curcumin may offer a promising therapeutic approach for metabolic diseases.The underlying mechanisms of curcumin are still necessary to be further explored and elucidated.

Fig.3. Autophagy-associated signal pathways of functional foods in chronic diseases.Resveratrol can activate autophagy via Sirt3,AMPK/Sirt1,AMPK/mTOR,JNK1/mTOR,and TyrRS/PARP1/Sirt1 signal pathways. EGCG can activate autophagy via AMPK, PI3K/NO and Nrf2 signal pathways. Curcumin can activate autophagy via AMPK/mTOR,PI3K/Akt/mTOR and FOXO1 signal pathways. Trehalose can activate autophagy via p38MAPK, FOXO1 and mTOR signal pathways. Therefore, the inducted autophagy is beneficial for the prevention and alleviation of chronic diseases.
5.4. Trehalose
Trehalose, a natural disaccharide, is widely produced in nonmammals such as fungi, yeasts, and similar organisms. It can maintain cell integrity by preventing protein denaturation [118].Since trehalose is safe at high concentrations, it has the potential of treating a series of chronic diseases. Pre-treatment with trehalose can induce autophagy and cell survival against H2O2in cell model. During trehalose treatment, the reduced levels of p62 and cleaved caspase-3 and the increased LC3-II/LC3-I ratio can be observed[119].In Akt2 knockout(Akt2-/-)mice,oral administration of trehalose can result in the increased LC3-II and decreased p62 and caspase-3.The enhanced phosphorylation of p38 mitogenactivated protein kinase (MAPK) and the up-regulation of FoxO1 can be detected in Akt2-/-mice, but can be suppressed by trehalose.
Meanwhile, the phosphorylation of Akt is unaffected by trehalose in Akt2-/-mice.in vitro,p38 MAPK and FoxO1 inhibitors can effectively decrease trehalose-offered beneficial contractile of cardiomyocytes under Akt2 ablation condition.Trehalose may rescue the reduced myocardial contractile due to the induced autophagy regulated by dephosphorylated p38 MAPK and FoxO1 [120].Macrophages play a critical role in clearing lipid and dead cell debris in atherosclerotic plaques.Trehalose,as an inducer of transcription factor EB (TFEB), regulates autophagy to promote the survival of macrophages in the highly pro-inflammatory and lipotoxic plaque environment. The macrophages induced by trehalose can suppress the hyperactivation of inflammasomes and the aggregation of cytotoxic proteins, and promote lipid degradation and deficient efferocytosis[121].Trehalose also can reduce atherosclerotic plaque burden for executing atheroprotective effects. Short-term intravenous administration of trehalose can reduce plaque grading and intima/media thickness ratio in high-cholesterol-fed rabbits[122]. Trehalose can reduce myocardial infarction (MI)-induced left ventricular (LV) dilation, cardiac remodeling and dysfunction through activating autophagy in a mouse model of chronic ischemic remodeling[123].Intraperitoneal injection of trehalose in Beclin1+/-mice, the up-regulated LC3-II in myocardial cells could be observed, suggesting the induced autophagy in heart in vivo[123]. An mTOR-independent autophagy can be activated by trehalose, which enhances the clearance of mutant huntingtin and mutants of α-synuclein associated with HD and PD.Both trehalose and rapamycin have the dual protective properties during the treatment of neurodegenerative diseases[124].Trehalose increases the clearance of abnormal proteins through inducing autophagy. In a PD mouse model with tauopathy,the treatment with 1%trehalose dissolved in drinking water can decrease the level of phosphorylated Tau protein [125], suggesting its neuroprotective effect(Fig.3).
5.5. Other natural products
Recently, other bioactive compounds in functional foods have also been confirmed to treat chronic diseases. Quercetin,a flavonoid, is known for its neuroprotective and antioxidant effects, and can significantly attenuate rotenone-induced behavioral impairment through augmenting autophagy and ameliorating ER stress-induced apoptosis [126]. Meanwhile, intracellular calcium storage may play an essential role in quercetin-induced autophagy[127].The expression of LC3-II is upregulated whereas p62 and mTOR are down-regulated by quercetin intervention,thereby accelerating degradation capacity of ox-LDL in a manner of autophagy[128].Therefore,quercetin can be used for the treatment of hyperlipidemia.
Dihydromyricetin(DHM),the bioactive component isolated and purified from Rattan tea, has been widely reported due to its functions of improving insulin sensitivity, and anti-diabetic, neuroprotective,cardioprotective,anti-inflammatory,and antioxidant properties, with the involved mechanisms associated with the regulation of AMPK, PGC-1α, MAPK, Akt, Nrf2 and NF-κB signal pathways[18,129–133].Similarly,DHM has the therapeutic potential in diabetic cardiomyopathy through activating autophagic signal pathway [130]. The Beclin1, Atg7, LC3-II/LC3-I ratio, and p-ULK1 reveal an obvious increase, and p62 exhibits a significant decrease upon DHM treatment of diabetes or diabetic cardiovascular diseases, thereby reducing oxidative stress and suppressing informatory factors such as IL-6 and TNF-α, as well as improving mitochondrial function. In addition, the glucose uptake in skeletal muscle can be enhanced during DHM intervention in vitro and in vivo,and the improvement of insulin sensitivity induced by DHM is distinctly reversed in the presence of autophagy inhibitor in C2C12 myotubes[129].Moreover,AMPK/PGC-1α/Sirt3 signal pathway also can be activated by DHM to induce autophagy, thereby improving insulin sensitivity in skeletal muscle[129].
6. Conclusion
Since functional foods reveal visible effects on chronice or metabolic diseases without any adverse impacts on the body,they will be regarded as the promising strategy for the prevention and natural supplementary therapy of these diseases as well as health promotion in the future.Up to the present,these autophagyassociated bioactive compounds in functional foods, such as RSV,EGCG, curcumin, trehalose or DHM for chronic or metabolic diseases have been systematically explored, which also provides a promising target based on autophagy signal pathways for the research and development of novel and effective natural products of functional foods in the future.
Conflicts of interest
These authors have declared no conflict of interest.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 31571228 and No. 31771318),Hubei Superior Discipline Groups of Physical Education and Health Promotion, and Outstanding Youth Scientific and Technological Innovation Team(T201624)from Hubei Provincial Department of Education,as well as Chutian Scholar Program and Innovative Startup Foundation from Wuhan Sports University to Ning Chen.
- 食品科学与人类健康(英文)的其它文章
- Microalgae:A potential alternative to health supplementation for humans
- Screening of potential GCMS derived antimigraine compound from the leaves of Abrus precatorius Linn to target“calcitonin gene related peptide”receptor using in silico analysis
- Rapid and easy determination of morphine in chafing dish condiments with colloidal gold labeling based lateral flow strips
- Optimization of process conditions for drying of catfish(Clarias gariepinus)using Response Surface Methodology(RSM)
- QSAR modeling of benzoquinone derivatives as 5-lipoxygenase inhibitors
- High uric acid model in Caenorhabditis elegans