
含苯环结构的糖类小分子胶凝剂的合成及其自组装行为研究
2019-05-24 02:02陈香李高莹莹步怀天
陕西科技大学学报 2019年3期
陈香李, 高莹莹, 杨 方, 步怀天
(陕西科技大学 化学与化工学院 教育部轻化工助剂化学与技术重点实验室, 陕西 西安 710021)
0 引言
自上世纪九十年代以来,小分子胶凝剂(Low Molecular Mass Gelators,LMMGs)形成的超分子凝胶及其自组装结构受到了广泛关注[1-4].超分子凝胶是由确定分子结构的小分子量有机化合物(M<3 000)通过分子间的相互作用(如氢键、色散力、范德华力、π-π堆积等)在溶剂中自组装形成三维网络结构,进而通过界面张力使溶剂失去宏观流动性而形成凝胶.相比于化学凝胶中三维网络间的共价键,超分子凝胶具有良好的溶胶-凝胶相转变可逆性,从而使得超分子凝胶具有对光、电、磁、热、pH、超声、氧化/还原等多重刺激响应性[5-8].
随着超分子凝胶的快速发展,功能性凝胶受到人们日益广泛的关注.研究LMMGs分子结构中的官能团在胶凝过程中发挥的作用,以及分析分子凝胶的形成机理,可以在LMMGs的分子设计中目的性的引入某些功能基团,诱导分子有效自组装形成凝胶,并赋予其特殊功能性.但是,胶凝剂分子的自组装并不是各官能团间的简单堆砌,各官能团之间通常具有协同作用或存在相互影响,胶凝剂自身的分子结构决定了其自组装行为.因此,研究胶凝剂的导向性合成仍是一个巨大挑战.
糖类化合物由于具有亲水性、手性骨架、良好的生物相容性等特点[9],已成功用于小分子胶凝剂的设计合成[10].糖类LMMGs由于结构的特殊性,在有机溶剂和水中具有丰富的自组装行为,表现出特殊的胶凝能力,所形成的凝胶在医学、生物学以及材料学等领域存在潜在的应用价值[11,12].但是,现阶段对糖类LMMGs的研究主要局限于吡喃型糖类,而对于其它类型(如直链型糖、呋喃型糖、五碳糖等)关注较少.此外,由于糖类LMMGs中分子间作用复杂且功能基团多样,目前很少研究胶凝剂分子的自组装行为与其分子结构和溶剂结构之间的相互关系.因此,设计合成结构更为新颖的糖类小分子、制备功能性糖类分子凝胶仍是这一领域的研究热点.
含芳环的糖类LMMGs利用芳环间的π-π堆积作用以及糖单元间的氢键作用,在溶剂中可能存在特殊的自组装行为.目前,以直链型糖类化合物为基元合成小分子胶凝剂的研究较少,而且对于含芳烃的糖类LMMGs的胶凝机理研究也不深入.因此,本文以(邻、间、对)苯二胺为连接臂将葡萄糖酸与胆固醇相连接,设计合成了3种含苯环结构的直链型糖类小分子化合物,研究了这些化合物在各类溶剂中的胶凝行为,通过SEM、FTIR、1H NMR、流变学、XRD等手段系统研究了小分子胶凝剂在溶剂中的自组装行为,提出了凝胶网络结构可能的形成机理.
1 实验部分
1.1 仪器及试剂
1.1.1 主要试剂
胆固醇氯甲酸酯(98%),邻苯二胺(AR, 97%),对苯二胺(AR, 97%),间苯二胺(99%):麦克林试剂,未经纯化直接使用;D-(+)-葡萄糖酸-δ-内酯(99%):阿拉丁试剂,未经纯化直接使用;实验所用溶剂均为国药集团化学试剂有限公司产品(分析纯),其中三乙胺(AR)用氢氧化钾除水后蒸馏纯化;四氢呋喃放入钠丝回流4 h后蒸馏后使用;二氯甲烷、三氯甲烷:经无水氯化钙干燥后蒸馏;甲醇、丙酮使用前经蒸馏纯化;实验用水经MilliQ超纯水机纯化.
1.1.2 主要仪器
瑞士Bruker公司AVANCE 400超导核磁共振仪(400 MHz,Me4Si为内标),德国Bruker Equinx55傅立叶变换红外光谱仪,德国Vario EL III型元素分析仪,美国UHR-TOF质谱仪(Bruker,ESI正离子光源),荷兰Philips-FEI公司Quanta 200扫描电子显微镜,美国TA公司AR-G2流变仪,日本理学公司的D/Max2550VB+/PC全自动XRD衍射仪.
1.2 胶凝剂CnG的合成与表征
中间产物Cn的合成参考文献[13]中的方法.CnG的合成路线示意图如图1所示.
C1G的合成:将中间体C1(0.52 g,1 mmol)和D-(+)-葡萄糖酸内酯(0.18 g,1 mmol)溶于40 mL甲醇中搅拌回流12 h.反应结束后,将反应液经抽滤后,所得到的固体用甲醇(3×100 mL)和水(3×100 mL)热洗以去除杂质,真空干燥后得到白色粉末状固体C1G,产率为62%.化合物C2G和C3G用同样方法合成.C2G为浅灰色固体粉末,C3G为浅粉色固体粉末.
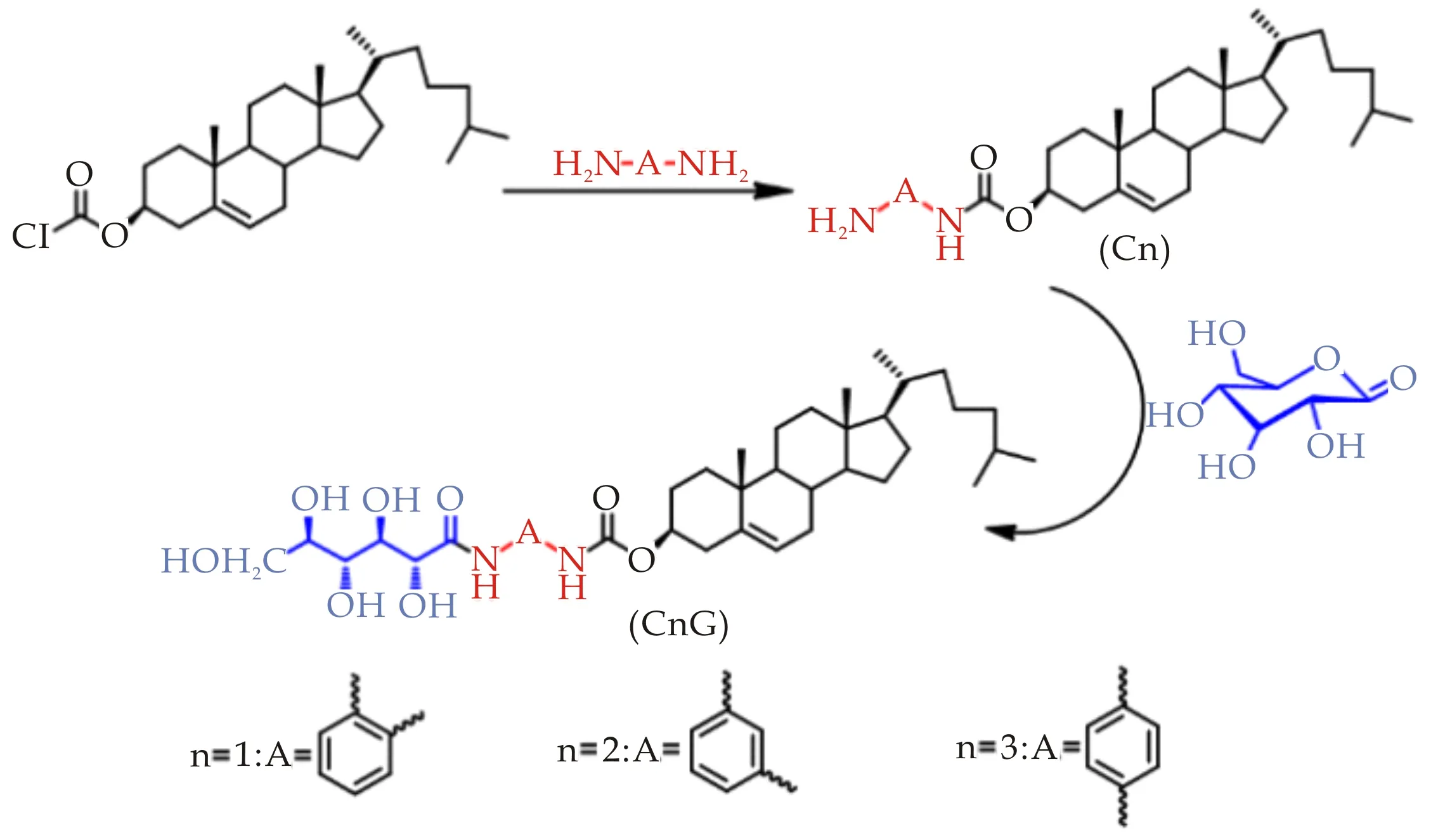
图1 化合物CnG的合成路线示意图
化合物C1G的分子结构表征如下:1H NMR(THF-d8/Me4Si,400 MHz):δ(ppm):8.11(1H,s,CONH),7.72~7.31(m,4H,benzene),7.2(1H,s,CONH),5.37(s,C=C,1H),4.50~3.30(m,11H,glucosylprotons),0.67~2.53(m,CH3,43H,cholesterylprotons).FTIR(υ/cm-1):3 315(-NH键的伸缩振动),1 726(C=O键的伸缩振动),1 630(C=C键的伸缩振动),1 539(-NH键的弯曲振动),1 235(-C-O-C-键的伸缩振动).元素分析C40H62N2O8计算值/%:C,68.74;H,8.94;N,4.01;O,18.31.实测值/%:C,68.71;H,9.02;N,4.12;O,18.36.MS(ESI):m/z计算值[M+Na]+:721.452 9,实测值:721.453 8.
化合物C2G的分子结构表征如下:1H NMR(THF-d8/Me4Si,400 MHz):δ(ppm):8.06(1H,s,CONH),7.81(1H,t,benzene),7.62~7.75(2H,s,benzenering),7.43~7.26(1H,s,benzenering),7.18(1H,s,CONH),5.47(s,C=C,1H),4.86~3.89(m,11H,glucosylprotons),0.68~2.43(m,CH3,43H,cholesterylprotons).FTIR(υ/cm-1):3 348(-NH键的伸缩振动),1 741(C=O键的伸缩振动),1 634(C=C键的伸缩振动),1 537(-NH键的弯曲振动),1 245(-C-O-C-键的伸缩振动).元素分析C40H62N2O8计算值/%:C,68.74;H,8.94;N,4.01;O,18.31.实测值/%:C,68.81;H,8.96;N,4.08;O,18.43.MS(ESI):m/z计算值[M+Na]+:721.452 9,实测值:721.453 5.
化合物C3G的分子结构表征如下:1H NMR(THF-d8/Me4Si,400 MHz):δ(ppm):8.09(1H,s,CONH),7.92~7.78(2H,d,benzenering),7.71~7.63(2H,d,benzene),7.59(1H,s,CONH),5.42(s,C=C,1H),4.53~3.38(m,11H,glucosylprotons),0.69~2.48(m,CH3,43H,cholesterylprotons).FTIR(υ/cm-1):3 329(-NH键的伸缩振动),1 753(C=O键的伸缩振动),1 648(C=C键的伸缩振动),1 541(-NH键的弯曲振动),1 241(-C-O-C-键的伸缩振动).元素分析C40H62N2O8计算值/%:C,68.74;H,8.94;N,4.01;O,18.31.实测值/%:C,68.79;H,9.03;N,4.22;O,18.46.MS(ESI):m/z计算值[M+Na]+:721.452 9,实测值:721.454 2.
1.3 胶凝试验
将0.025 g胶凝剂置于带盖样品瓶(d≈10 mm)中,加入1.0 mL溶剂密封静置24 h.室温下为溶液或加热冷却至室温后仍为溶液,记为“S”,室温下完全溶解,静置后形成凝胶,记为“Grt”;加热溶解,冷却至室温静置后形成凝胶,记为“G”;若形成透明凝胶,记为“TG”;若经过超声诱导后形成凝胶,记为“G*”;加热后在冷却过程中析出沉淀,记为“P”;,记为“S”;冷却后为粘稠溶液,记为“VS”;加热过程中基本不溶,记为“I”;冷却过程中部分形成凝胶,记为“PG”.
1.4 理论模拟计算
本实验使用Materials Studio 7.0软件进行分子模拟计算.首先在Visualizer模块中分别构建胶凝剂分子C1G、C2G和C3G的单体模型,使用Forcite模块中的Geometry Optimization功能分别对所构建的单体模型进行能量最小化,能量最小化算法选择Smart minimizer,体系的能量趋于最小值,即化合物的空间构象达到了最稳定[14].将模拟计算后所得到的分子模型测量长度(两端原子中心之间的距离加上两端原子的范德华半径即为胶凝剂分子的拉伸长度).
2 结果与讨论
2.1 胶凝行为
考察了胶凝剂分子CnG(n=1,2,3)在30种溶剂中的胶凝行为,样品浓度为2.5%(w/v),实验结果如表1所示.由表1可知,胶凝剂C2G的胶凝能力明显优于胶凝剂C1G和C3G,C1G能够胶凝甲苯和水两种溶剂,C3G只能胶凝DMSO一种溶剂,而C2G可以胶凝水、DMSO、甲苯、正丁醇、正戊醇、正己醇、正庚醇、正辛醇、正壬醇、正癸醇十种溶剂,且在DMSO中能够室温成胶.
究其原因,可分析其分子结构,胶凝剂C2G分子中的苯环是通过间位与其它基团连接,而C1G和C3G的分子结构中的苯环分别通过邻位和对位与其它基团连接,模拟计算这三种化合物在能量最低时的空间构型如图2所示,可以看出C1G分子的空间构型出现明显弯折(如图2(a)所示),这使得分子间堆积时出现了较大位阻,从而阻碍了胶凝剂的簇集;C3G的空间构型(如图2(c)所示)中的糖结构部分出现非常强的分子内聚集,这也在一定程度上阻碍了胶凝剂的簇集;而C2G的空间构型较为舒展,其位阻较小(如图2(b)所示),明显具有更合理的分子间堆积趋势.由此也证明了胶凝剂分子通过苯环间位连接其他基团时能够使更多溶剂胶凝的原因.同时,从胶凝表中可以明显看出,溶剂极性对胶凝剂的胶凝行为具有明显影响,可以看出具有较好胶凝能力的C2G在极性较大的溶剂(如醇类)中更容易成胶,这也归结于胶凝剂分子结构中含有葡萄糖酸残基,富含羟基基团,可以更容易与质子性溶剂发生氢键结合,更有利于三维网络结构的生成,进而促进成胶.
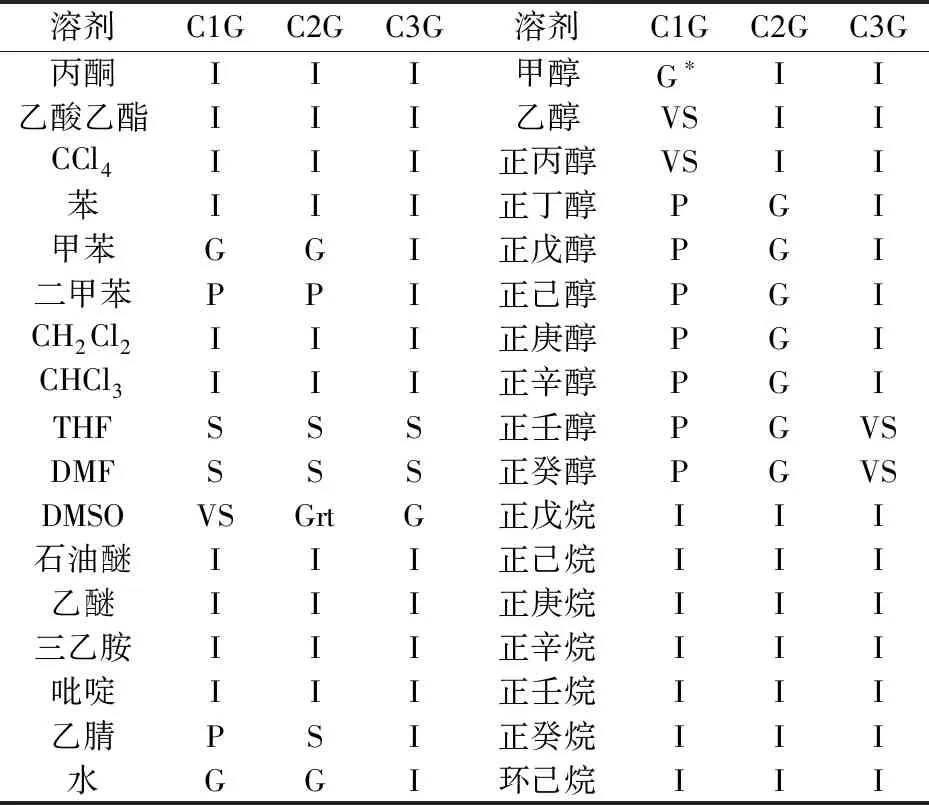
表1 化合物CnG在不同溶剂中的
2.2 凝胶的微观结构
根据上述分析,我们选择能够室温胶凝的C2G/DMSO凝胶体系为研究对象,深入研究其胶凝行为及其成胶机理.
利用环境扫描电镜观察了不同浓度的C2G/DMSO凝胶体系干凝胶的微观形貌,如图3所示.可以看出,胶凝剂分子C2G在DMSO中,当浓度为1.0%(w/v)以下,凝胶的聚集结构以纤维状存在,当浓度进一步增大后,凝胶结构逐渐变为纤维与球状结构并存的形貌,而当浓度增加到2.5%(w/v)时,凝胶的微观结构则呈现为球状聚集体.


(a)0.5 %(b)1.0 %(c)1.5 %(d)2.5 %图3 不同浓度的C2G/DMSO凝胶体系干凝胶的扫描电镜照片
为了进一步研究C2G在DMSO中的聚集过程.将浓度调至0.01%(w/v),此时C2G在DMSO中溶解(C2G/DMSO为超级胶凝体系,其最低胶凝浓度(CGC)为0.06%(w/v)),但从该体系的AFM照片(如图4所示)可以看出,在此浓度下,C2G分子在DMSO中已经开始聚集,缠绕形成一股具有左手螺旋的纤维.这表明,随着胶凝剂浓度的增加,凝胶的微观结构逐渐由纤维结构演化成为球状结构.
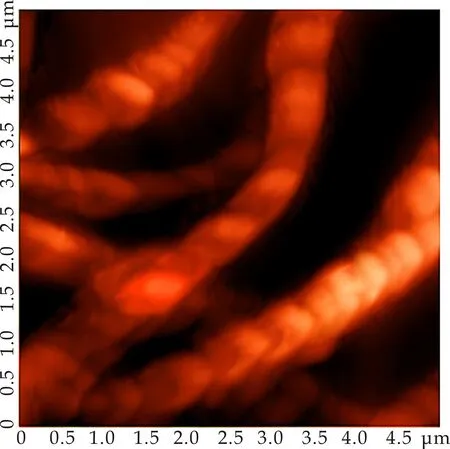
图4 0.01%的C2G/DMSO体系的AFM照片
2.3 凝胶的流变学
在研究过程中发现,C2G在室温下能够使DMSO胶凝,并能通过加热-冷却循环或者机械摇晃-停止循环使C2G/DMSO体系出现凝胶-溶胶相转变过程,表现出良好的剪切触变性,通过测量 C2G/DMSO体系的流变学性质科学解释该凝胶体系的这一现象.
测量不同浓度下C2G/DMSO凝胶体系的储能模量G′随剪切应力σ的变化趋势,如图5所示,发现当胶凝剂C2G的浓度从1.0%增加到2.5%(w/v)时,凝胶体系的G′值从81.42 Pa 逐渐增加到4124.02 Pa,其相应的屈服应力从21.92 Pa 增加到175.68 Pa.这表明凝胶的流变学性质(机械强度和稳定性)受体系中胶凝剂浓度的影响较大,随着体系中胶凝剂浓度的增大而加强.

图5 不同浓度的C2G/DMSO凝胶体系的储能模量G′随剪切应力σ的变化曲线图
流变测试中的频率扫描是检测体系对外界刺激忍受能力的一种常用方法[15].选择C2G在DMSO中浓度为2.5%(w/v)的凝胶体系,取其线性范围内的剪切力60.0 Pa,测量结果如图6所示.可以看出,当角频率在1.0 rad s-1到 628 rad s-1范围内,储能模量G′一直大于损耗模量G″,表现为凝胶的固体属性[16, 17].且G′和G″的数值没有发生大范围波动,这说明该凝胶体系对外界应力刺激具有很好的忍受能力,表现为典型的黏弹性流变学性质[18, 19].
为了研究该凝胶体系的触变性和可恢复性,测试了浓度为2.5%(w/v)的C2G/DMSO凝胶体系经剪切应力扫描破坏后随时间恢复变化的情况.经剪切应变扫描后使凝胶破坏后,撤去剪切力,立刻开始监测该体系的G′随时间的变化(该过程为了防止机械力过大而影响凝胶的恢复过程,监测时取较小的剪切力5.0 Pa,角频率为6.28 rad/s).取一次剪切应变扫描和一次恢复性扫描作为一个循环单元,对其进行了6次重复测量,结果如图7所示,每个循环中,在撤去外力后体系很快就恢复到被破坏前的数值.并且测量至第6个循环,所得到的G′数据与第一次测量数据相比变化并不大,说明该体系具有可重复剪切触变性[20-22].

图6 C2G/DMSO凝胶体系的G′和G″随频率的变化曲线图
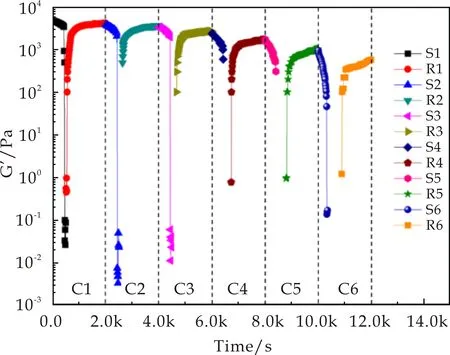
图7 C2G/DMSO凝胶的G′随时间变化曲线图
2.4 成胶机理
2.4.1 红外光谱
FTIR光谱是证实体系中是否存在分子间氢键的有效方法[23].图8给出了C2G/THF溶液和C2G/DMSO凝胶的FTIR光谱图.C2G在THF能够形成真溶液,即C2G在THF中是以单分子态存在,其FTIR光谱如图8中a曲线所示,3 348 cm-1和1 537 cm-1处分别对应N-H键的伸缩振动以及弯曲振动的特征吸收.1 741 cm-1和1 664 cm-1处分别对应于酯基中的C=O伸缩振动和酰胺中的C=O伸缩振动.但在C2G/DMSO干凝胶的FTIR图(如图8中b曲线所示),可以明显看到这几个红外特征峰位移发生了明显移动,分别移动至3 216 cm-1、1 732 cm-1、1 637 cm-1和1 526 cm-1,表明凝胶体系中存在分子间氢键作用,此为凝胶形成的主要驱动力之一[24-27].
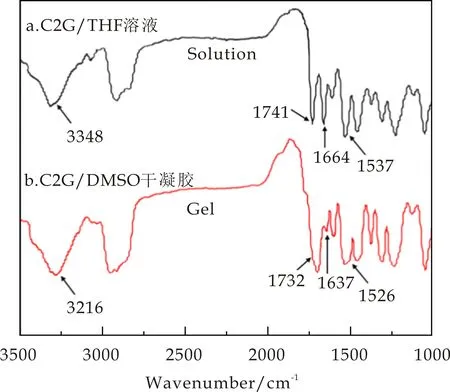
图8 C2G/THF溶液和C2G/DMSO干凝胶的红外光图谱
2.4.2 核磁共振谱
利用1H NMR技术可以为氢键作用和苯环的π-π堆积作用提供有效证据,能够进一步研究凝胶网络结构的形成机理,深入研究溶液中胶凝剂分子的聚集行为[28, 29].选用胶凝剂的良溶剂氘代四氢呋喃作为溶剂,图9(a)为2%(w/v)的C2G/THF在不同温度下的核磁图谱.可以看出,当温度从298 K升至338 K时,两个-NH的化学位移分别从7.18 ppm和8.06 ppm移动至7.07 ppm和7.99 ppm,而归属于葡萄糖酸残基的-OH的化学位移也从4.86 ppm移动至4.69 ppm.显然,这说明了胶凝剂分子中的-NH和-OH参与了氢键形成,与FTIR测试结果一致.
同时,增加了C2G的浓度,测试其在氘代四氢呋喃中不同浓度的1H NMR,如图9(b)所示.发现-NH和-OH的化学位移均向低场发生了相应移动,说明在该体系中存在分子间氢键作用[30].并且,胆固醇片段上特征氢的化学位移(5.47 ppm)和苯环上氢的化学位移(7.81 ppm)无论是在变温核磁还是在变浓度核磁中,均能看到发生了轻微的移动,这表明胶凝剂分子的胆固醇片段与苯环具有较弱的π-π堆积作用[12, 31].综上所述,氢键和π-π堆积作用凝胶形成的主要驱动力.
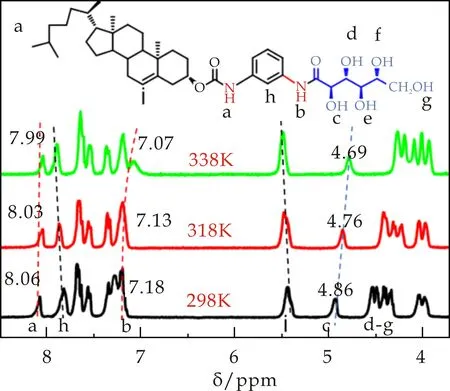
(a)C2G/THF-d8在不同温度(2% w/v)的核磁图谱片段

(b)C2G/THF-d8在不同浓度下(2% w/v)的核磁图谱片段图9 C2G/THF-d8的变温和变浓度核磁图谱片段
2.4.3 XRD
通过测定X-射线衍射可以明确胶凝剂分子在凝胶中的堆积方式,进一步提出合理的胶凝机理和堆积[32-35].测定了胶凝剂C2G的粉末态及其在DMSO中形成干凝胶的XRD图谱,结果如图10所示.可以看出,无论是粉末态还是凝胶态均出现了三个明显的衍射峰,凝胶态对应的d值分别为4.12、2.12、1.38,而粉末态对应的d值分别为3.94、1.99、1.33,两种状态的d值比例均接近 1∶1/2∶1/3,这表明胶凝剂分子的粉末态和在C2G/DMSO凝胶中具有相同的分子堆积模式,均呈片层状堆积,层间距是3.98 nm,这与模拟计算的两个单分子形成的分子单元长度接近,如图10插图所示.
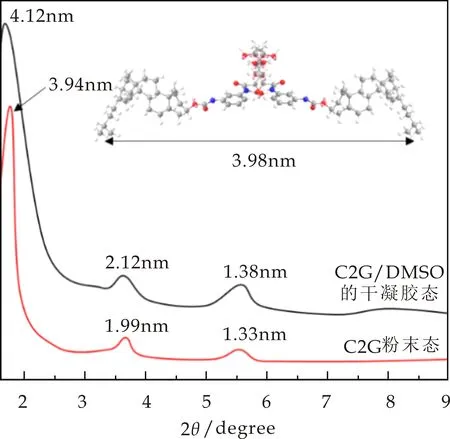
图10 C2G/DMSO的干凝胶态和C2G粉末态的XRD图谱
2.4.4 堆积模型
综上所述,提出了C2G在DMSO中凝胶网络结构可能的自组装过程及机理如图11所示.即葡萄糖酸残基的-OH间以及连接臂上-NH与C=O间的分子间氢键作用、苯环的π-π堆积作用和胆固醇之间的范德华相互作用是凝胶网络形成和维系的主要驱动力.这两种弱相互作用相互协同使胶凝剂分子先聚集成左右螺旋状原纤维,进而相互交织形成球状结构,最后将溶剂固定形成凝胶.
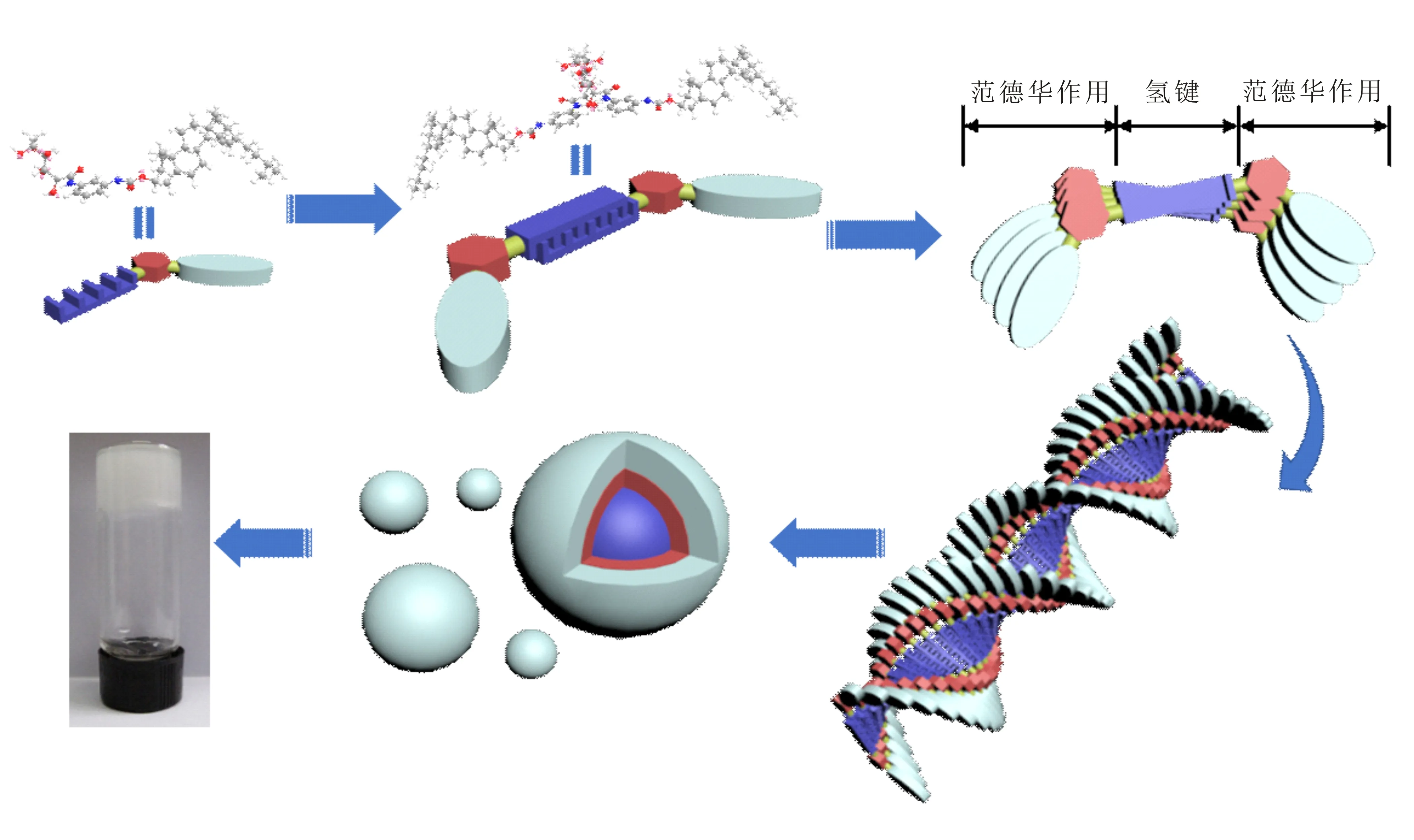
图11 C2G/DMSO体系的聚集体形成机理示意图
3 结论
本文系统考察了3种含苯环的糖类小分子胶凝剂在常见溶剂中的胶凝行为.结果表明,胶凝剂分子结构中苯环连接方式不同,其空间位阻不同,进而使得3种化合物的胶凝能力存在明显差异.间位连接的胶凝剂分子C2G的胶凝能力明显优于邻位和间位连接的胶凝剂分子C1G和C3G,且C2G在DMSO中最低胶凝浓度可达0.06%(w/v).
选取胶凝能力较好的C2G进行深入研究,发现溶剂本性和胶凝剂浓度对胶凝剂在溶剂中的胶凝行为、聚集体结构和流变学性能具有显著影响;流变学研究表明C2G/DMSO具有较好的机械稳定性和剪切触变性.FTIR和变温、变浓度1H NMR表明氢键及π-π堆积作用是该类超分子凝胶形成的两大驱动力.XRD结果表明胶凝剂C2G在DMSO中以层状模型堆积.根据这些测试结果,提出了C2G在DMSO中可能的堆积模型.
猜你喜欢
油田化学(2022年4期)2023-01-10
中学化学(2022年5期)2022-06-17
华南师范大学学报(自然科学版)(2021年4期)2021-08-30
化学与生物工程(2021年1期)2021-01-22
化工技术与开发(2020年6期)2020-06-24
理科考试研究·高中(2019年8期)2019-09-19
天然产物研究与开发(2018年6期)2018-07-09
家庭用药(2018年11期)2018-01-23
中国民族医药杂志(2016年7期)2016-05-09
化学教学(2015年11期)2015-12-19
