
高纯度亚麻木酚素的分离纯化和分析
2019-05-21 11:59贺天雨赵新颖席兴军张经华
食品科学 2019年8期
王 尉,贺天雨,赵新颖,*,席兴军,兰 韬,杜 宁,张经华
(1.北京市理化分析测试中心,北京 100089;2.中国标准化研究院,北京 100191)
亚麻(Linum usitatissimum L.)又称胡麻,与花生、大豆等同属重要的油料作物[1],其种子、种皮中木酚素含量最高,约为其他植物的75~800 倍[2]。同时,亚麻还是重要的纤维制造原料之一,因此作为重要的生产和生活材料,人们对于亚麻的研究十分重视且从未间断。自1956年Bakke等[3]首次将亚麻木酚素从亚麻籽中分离出来,科技工作者便对亚麻的药用价值进行了系统深入的研究。据报道,亚麻木酚素具有较强的抗氧化活性[4-6]和抗炎作用[7],对糖尿病[8-9]、心血管疾病[10-12]、肾脏病[13-14],尤其是对于乳腺癌[15-20]、经期综合征、骨质疏松[21-22]等雌激素依赖性疾病有较好的预防作用。近年来,亚麻木酚素提取物产品已经广泛用于胶囊压片、谷物早餐和快餐食品的添加剂[23]。因此,开发高效、简单的高纯度亚麻木酚素分离分析技术对于充分发挥亚麻的应用价值具有重要的意义。
目前,亚麻木酚素的制备工艺多集中于提取方法及大孔吸附树脂纯化等方面的研究[24-27],其制备所得的亚麻木酚素纯度不高,鲜有高纯度亚麻木酚素分离纯化的报道。此外,亚麻木酚素的纯度或含量的分析方法也仅限于单一条件的高效液相色谱(high performance liquid chromatography,HPLC)法或紫外分光光度法等[1],并不能全面、准确反映样品中杂质含量的情况。本研究通过正己烷脱脂、乙醇提取、大孔吸附树脂初步分离、高速逆流色谱(high-speed countercurrent chromatography,HSCCC)纯化等方法制备得到高纯度的亚麻木酚素,方法简单可行;综合利用薄层色谱(thin layer chromatography,TLC)以及多种HPLC条件和联用技术进行纯度分析,结果准确可靠。最后通过紫外光谱(ultraviolet spectrum,UV)、红外光谱(infrared spectrum,IR)、高分辨质谱(high-resolution mass spectrometry,HRMS)、核磁共振波谱(nuclear magnetic resonance spectroscopy,NMR)和元素分析等方法对亚麻木酚素进行结构鉴定。
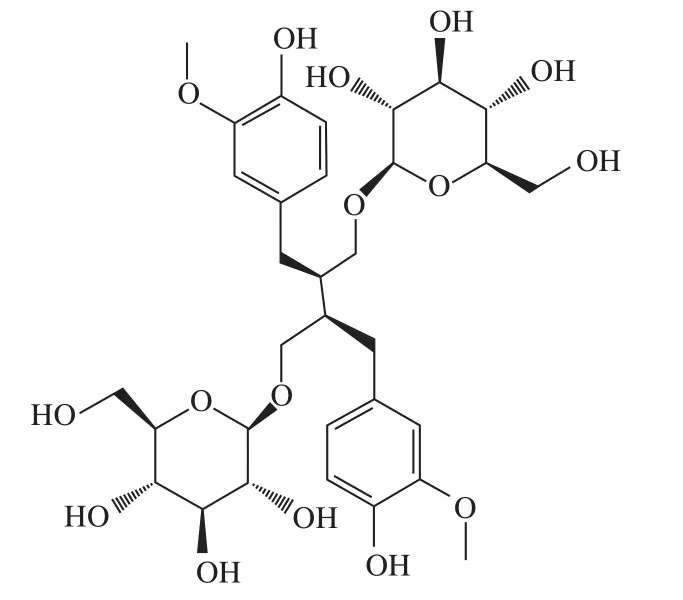
图 1 亚麻木酚素的化学结构式Fig. 1 Chemical formula of flax lignan
1 材料与方法
1.1 材料与试剂
干燥亚麻籽 市售。
正己烷、乙醇、叔丁基甲醚、正丁醇、氯仿、乙腈、氢氧化钠、盐酸、硫酸(均为分析纯) 国药集团化学试剂有限公司;乙腈(色谱纯) 美国Fisher Scientific科技公司。
1.2 仪器与设备
TBE-300B高速逆流色谱仪 上海同田生物技术有限公司;LC-20A HPLC系统(配SPD-M20A和ELSD-LTII检测器)、UV-1800紫外-可见分光光度计 日本岛津公司;PerkinElmer spectrum 400傅里叶变换红外-近红外光谱仪 美国珀金埃尔默公司;Q Exactive Orbitrap质谱仪美国Thermo公司;DD2 600 MHz超导核磁共振谱仪美国Aglient公司;Vario EL III全自动元素分析仪 德国Elementar公司。
1.3 方法
1.3.1 亚麻木酚素的提取及分离纯化
1.3.1.1 样品的提取
将亚麻籽粉碎后过40 目筛,称取100.0 g按照料液比1∶20(g/mL)加入正己烷,室温浸泡脱脂6 h。对脱脂后的样品按料液比1∶20(g/mL)加入乙醇,超声波辅助提取3 次,离心分离,合并上清液。在上清液中加入6 mol/L NaOH溶液至最终NaOH浓度为0.25 mol/L,室温碱解2 h后,加入6 mol/L HCl溶液中和至pH 4.0,旋转蒸发去除乙醇,可得亚麻籽水解物9.6 g。
1.3.1.2 分离纯化
首先,采用AB-8大孔吸附树脂对亚麻籽水解物进行初步分离,依次使用蒸馏水和80%乙醇溶液洗脱,收集80%乙醇溶液洗脱产物,旋转蒸发去除溶剂,可得亚麻籽初步分离样品105 mg。然后,采用HSCCC对该样品进行纯化,溶剂体系为叔丁基甲醚-正丁醇-乙腈-水(1∶3∶1∶5,V/V),转速900 r/min,流速1.2 mL/min,分离温度25 ℃,检测波长280 nm。根据HSCCC图谱收集目标化合物,旋转蒸发去除溶剂,冷冻干燥后得到高纯度亚麻木酚素样品58 mg。
1.3.2 纯度分析
1.3.2.1 TLC纯度分析
分别采用两种展开条件对亚麻籽提取物和亚麻木酚素进行TLC纯度分析,展开剂分别为乙酸乙酯-甲醇-水-甲酸(77∶13∶10∶5,V/V)和氯仿-甲醇-乙酸(8∶4∶0.5,V/V),显色剂为5%硫酸-乙醇溶液,喷洒后于105 ℃加热显色。
1.3.2.2 多种HPLC条件纯度分析
在流动相A为乙腈,B为1%甲酸,流速1.0 mL/min、柱温35 ℃条件下,对HPLC洗脱条件、不同色谱柱、高效液相色谱-二极管阵列检测器-蒸发光散射检测器(high performance liquid chromatography-diode array detectorevaporative light scattering detector,HPLC-DAD-ELSD)联用等方法对亚麻木酚素进行纯度分析,并采用峰面积归一化法计算纯度。
1.3.2.3 HPLC-MS联用纯度分析
采用HPLC-MS的正、负离子模式对亚麻木酚素进行纯度分析。HPLC条件:色谱柱:ACQUITY UPLC(2.1 mm×100 mm,1.7 μm);流动相:A为乙腈,B为0.3%甲酸,0~10 min,20% A;流速:0.2 mL/min;柱温:35 ℃;运行时间:10 min。MS条件:锥孔气流速40 L/min;毛细管电压3.0 kV;脱溶剂温度320 ℃;质量扫描范围m/z 150~2 000。
1.3.3 结构鉴定
对分离纯化后的样品通过UV、IR、HRMS、NMR和元素分析进行结构鉴定。UV分析条件:甲醇作为溶剂,扫描范围200~400 nm;IR分析条件:KBr压片法,扫描范围400~4 000 cm-1;MS分析条件同HPLC-MS联用纯度分析;NMR分析条件:氘代溶剂为氘代甲醇(CD3OD),采集13C-NMR和1H-NMR图。
2 结果与分析
2.1 亚麻木酚素的提取及分离纯化
首先,对粉碎后亚麻籽样品采用正己烷脱脂、乙醇超声波辅助提取得到亚麻籽提取物样品。由于亚麻籽中部分亚麻木酚素会与3-羟基-3-甲基-戊二酸形成络合物[28-29],为提高提取效率,采用碱水解[30-31]的方式有利于释放更多亚麻木酚素成分,故通过NaOH碱水解,HCl中和后制备得到亚麻籽水解物。然后,对该样品采用AB-8大孔吸附树脂初步分离,收集80%乙醇洗脱产物得到亚麻籽初步分离样品。最后,采用HSCCC叔丁基甲醚-正丁醇-乙腈-水(1∶3∶1∶5,V/V)溶剂体系纯化得到亚麻木酚素样品(图2),对以上得到的亚麻籽提取物、亚麻籽初步分离、亚麻木酚素3 个样品经HPLC分析,采用峰面积归一法计算纯度,其纯度分别为8.7%、90.1%、99.4%(图3)。
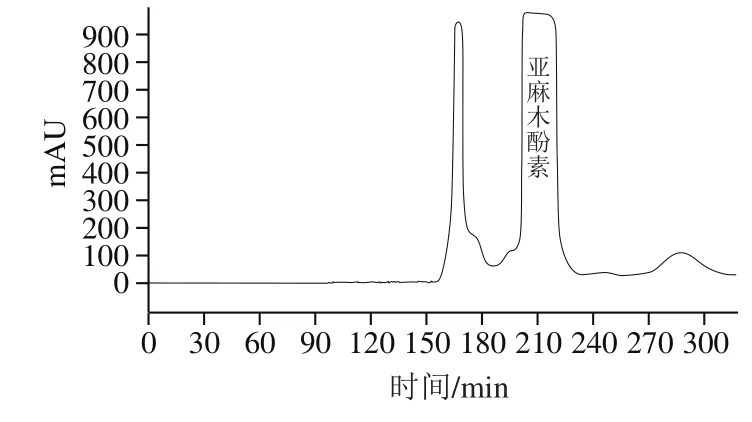
图 2 亚麻木酚素的HSCCC图Fig. 2 HSCCC chromatogram of flax lignan

图 3 亚麻木酚素的HPLC图Fig. 3 HPLC chromatograms of flax lignan
2.2 纯度分析
2.2.1 TLC纯度分析
分别采用两种展开条件对亚麻籽提取物(图4,点样位置1)和3 种浓度样品亚麻木酚素(图4,点样位置2、3、4)进行TLC纯度分析。展开剂为乙酸乙酯-甲醇-水-甲酸(77∶13∶10∶5,V/V)的Rf值为0.36,展开剂为氯仿-甲醇-乙酸(8∶4∶0.5,V/V)的Rf值为0.60,通过实验结果可知,未在TLC谱图中发现其他杂质斑点,表明分离纯化所得亚麻木酚素的纯度较高。
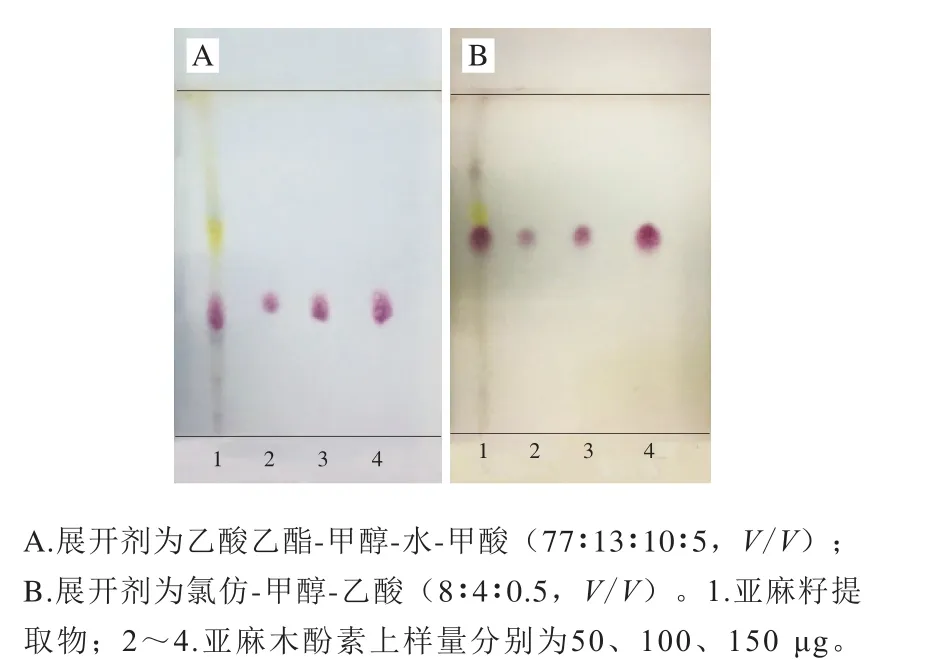
图 4 亚麻木酚素的TLC图Fig. 4 TLC analysis of flax lignan
2.2.2 多种HPLC条件纯度分析
为全面反映杂质的情况,需要采用多种HPLC条件对目标物进行分析。本实验比较恒定洗脱、梯度洗脱、色谱柱类型、HPLC-DAD-ELSD联用的方法,并采用峰面积归一化法对亚麻木酚素进行纯度分析,结果见表1。实验结果表明,多种方法测得的目标物纯度基本一致,均高于99%,其中HPLC-DAD-ELSD联用谱图见图5、6。

表 1 不同HPLC条件分析结果Table 1 Results of HPLC analysis under different conditions

图 5 亚麻木酚素的DAD谱图Fig. 5 DAD spectrum of flax lignan
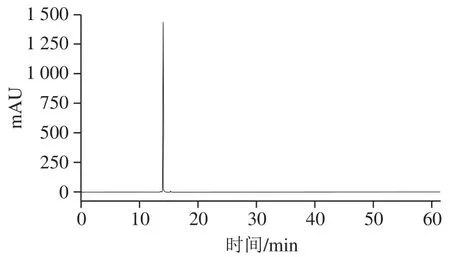
图 6 亚麻木酚素的ELSD色谱图Fig. 6 ELSD chromatogram of flax lignan
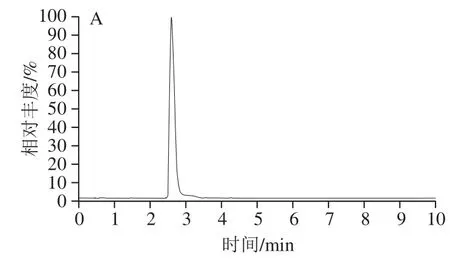

图 7 亚麻木酚素的总离子流色谱图Fig. 7 Total ion current chromatograms of flax lignan
2.2.3 HPLC-MS联用纯度分析采用正、负离子两种模式对亚麻木酚素进行纯度分析。通过实验结果(图7)可知,未在总离子流谱图中发现其他杂质峰存在。
2.3 结构鉴定结果

表 2 亚麻木酚素的1H-NMR数据Table 2 1H-NMR data of flax lignan

表 3 亚麻木酚素的13C-NMR数据Table 3 13C-NMR data of flax lignan
分离纯化后样品经元素分析结果显示:C:56.2%,H:6.5%,与亚麻木酚素的元素组成计算值(C:56.0%,H:6.7%)相符。UV最大吸收波长为281 nm(甲醇溶剂);IR吸收峰为3 406 cm-1(-OH伸缩振动)、2 929 cm-1(C-H伸缩振动)、1 604、1 516、1 430 cm-1(芳环骨架振动)、1 272、1 076、1 031 cm-1(C-O伸缩振动)(图8),UV、IR数据与文献[31]比较,光谱特征一致。如图9所示,HRMS正离子模式给出m/z 709.266 1[M+Na]+(C32H46O16Na精确分子质量计算值:m/z 709.268 4),m/z 327.158 4[M-2glu-2H2O+H]+(C20H23O4精确分子质量计算值:m/z 327.159 6),负离子模式给出m/z 731.276 7[M+COOH]-(C33H47O18精确分子质量计算值:m/z 731.276 2),m/z 685.270 6[M-H]-(C32H45O16精确分子质量计算值:m/z 685.270 8),以上数据均与亚麻木酚素精确分子质量相符。通过1H-NMR和13C-NMR鉴定(表2、3和图10),并与文献[32-33]比较,其核磁谱数据与报道的亚麻木酚素化合物一致,确定该样品为亚麻木酚素。
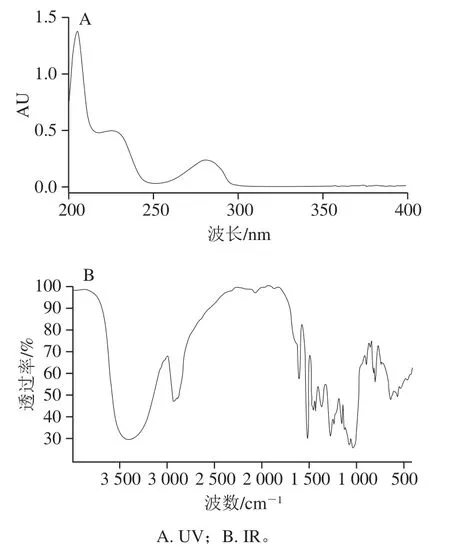
图 8 亚麻木酚素的UV和IR的光谱图Fig. 8 UV and IR spectra of flax lignan

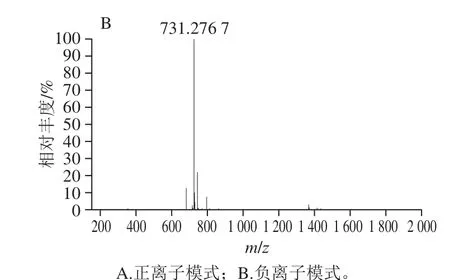
图 9 亚麻木酚素的HRMS图Fig. 9 HRMS spectra of flax lignan
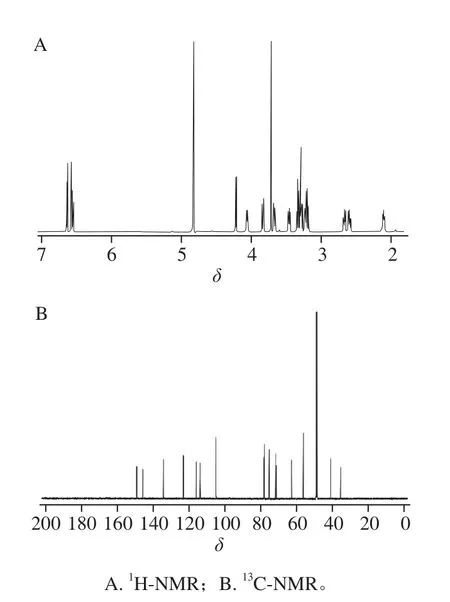
图 10 亚麻木酚素的NMR图Fig. 10 NMR spectra of flax lignan
3 结 论
本实验建立了从亚麻籽中分离纯化亚麻木酚素的方法,通过大孔吸附树脂和HSCCC两步分离纯化即可得到纯度在99.3%~99.5%之间高纯度亚麻木酚素,方法简单可行。这将有效促进亚麻药理活性的深入研究,推动亚麻木酚素对照品的广泛研制,提升亚麻相关产品的质量品质,进一步提高亚麻作物的经济价值。同时,本研究所采用的多种分析技术组合的纯度分析方法,也对制备其他高纯度天然产物单体具有一定的借鉴作用。
猜你喜欢
中华诗词(2022年6期)2022-12-31
上海化工(2021年1期)2021-02-26
陶瓷学报(2019年6期)2019-10-27
当代陕西(2019年16期)2019-09-25
中国麻业科学(2018年6期)2018-04-09
中国宝玉石(2018年6期)2018-03-05
鹿鸣(2018年1期)2018-01-30
散文诗世界(2017年3期)2017-11-13
大陆桥视野·下(2016年9期)2017-05-08
南方文学(2016年3期)2016-06-12
