
基于多重PCR 靶向二代测序的近交系小鼠遗传质量监测方法建立
2019-05-17 08:43王亚恒周宇荀肖君华鲍世民
实验动物与比较医学 2019年2期
钱 强, 徐 园, 王亚恒, 周宇荀, 肖君华, 韩 琳, 鲍世民, 李 凯
(1. 东华大学化学化工与生物工程学院, 上海 201620;2. 上海市第十人民医院, 上海 200072;3.中国科学院上海生命科学院, 上海 200031)
动物遗传质量对实验结果有重要影响。近交系小鼠是医学生物学研究中广泛应用的实验动物, 其本身的质量尤其是个体基因的纯合性与不同个体遗传一致性对实验结果的可比性、可重复性和准确性起着决定性作用。Taft 等[1]研究表明, 即便少量的遗传污染也能造成结果重复性困扰, 故有必要确认品系的遗传背景以及是否发生基因突变和遗传污染。
由于小鼠基因组中单核苷酸多态性(SNP)位点数量庞大,且比短串联重复序列(STR)易于实现高通量基因分型,故国际上较知名的实验动物公司均已使用SNP 分型技术进行遗传检测。如2004 年美国 Jackson 实验室筛选出28 个SNP 位点,用以鉴别48 种近交系小鼠[2],他们额外提供2 000 个SNP的芯片(http://jaxservices.jax.org /genome /snp.html)用于检测与基因定位;Charles River 实验室目前采用32个位点和384个位点的两个SNP组合对小鼠进行遗传检测(http://www.criver.com); Taconic 实验室采用96 个位点的SNP 组合对小鼠进行遗传检测;Harlan 实验室采用48 个位点的SNP 组合对小鼠进行遗传检测。可见,基于SNP 小鼠遗传质量检测方法逐渐成为国际通行标准。
基于多重PCR的SNP分型方案, 是一种高通量与高特异性的SNP分型方案[3]。本研究在PCR-LDR(连接酶检测反应)技术的SNP分型方案被用于小鼠遗传鉴定的基础上[4],筛选出染色体上均匀分布的112 个SNP 位点,利用靶向建库测序技术,以期实现小鼠遗传质量检测的高通量SNP 分型方案。
1 材料与方法
1.1 实验动物
本实验小鼠DNA 源于斯莱克实验动物有限责任公司[SCXK(沪)2012-0002], 共有4 批小鼠样本(4批样本数量分别为16、43、6 和60; 有10 个近交系品系分别为C57BL/6、FVB、C3H/He、BALB/c、DBA/2、DBA/1、AKR、CBA、SJL/J、NOD)。动物实验遵守《实验动物管理条例》。收集这些小鼠尾组织,-20 ℃保存备用。
1.2 主要仪器和试剂
PCR 仪(A-100),购自杭州朗基科学仪器有限公司; PCR 仪(Gene Amp PCR system 9600),购自美国Norwalk 公司; 电泳仪(JY600+),购自北京君意东方电泳设备有限公司; 全自动紫外与可见分析装置(FR-200A)、生物电泳图像分析系统,均购自上海复日科技有限公司。PCR 引物(PAGE 纯化)购自上海百力格生物技术有限公司; dNTP(promega)购自上海有渔生物工程有限公司; Taq酶体系和ddH2O属于实验室自制的。
1.3 SNP 位点筛选
从NCBI数据库查找在小鼠品系间中存在较高多态性的SNP 位点,为了避免连锁的可能性和提高分辨率,保证结果准确可靠,位点筛选原则如下: (1)尽量选择分布于不同染色体上,包括所有的常染色体与X 染色体,每条染色体所含SNP 最少为3 个,最多为8 个,Y 染色体因为雄性小鼠独有,且多样性极低,故未选; (2)尽量选择品系间差异大的SNP 位点; 我们最终选择了112 个SNP 位点(图1)。
1.4 小鼠全基因组DNA 抽提
使用动物基因组DNA 快速提取试剂盒Tiangen(天根生化科技有限公司),从小鼠尾尖抽取全基因组DNA。吸取1 μL 抽提好的DNA,在1%琼脂糖凝胶电泳中检测其浓度,然后将所有的DNA 样本标化到浓度为30 ng/μL。-20 ℃储存备用。
1.5 引物设计

图1 112 个SNP 位点在染色体上的分布图Figure 1 Distribution of 112 SNP loci on chromosomes
从NCBI数据库下载含有112 个SNP基因位点的靶向区域的序列。为了获得特异性PCR 产物,设计了含有靶序列和通用序列的嵌合特异性引物。PCR 反应的产物大小在200~250 bp,引物长度为20~30 bp,熔解温度(Tm)为55~65 ℃,GC 含量为20%~80%。为了区分不同的样品, 我们设计了96 对含有索引序列和通用序列的条形码引物(图2)。最后使用Illumina 公司的P5 与P7 引物,统一建库。
1.6 多重PCR 建库

图2 多重PCR 的设计Figure 2 Design of multiplex PCR
第一轮靶向SNP位点的特异性扩增体系为10 μL,含1 μL 小鼠基因组DNA(15~20 ng/μL),1 μL 1×PCR 缓冲液(含15 mmol/μL Mg2+),1 μL 1×PCR辅助剂,1 μL 200 μmol/μL dNTPs,1 μL 引物(0.2 μmol/μL), 0.6 μL 25 mmol/μL Mg2+, 0.1 μL 热启动DNA 聚合酶(5U/μL), 再加重蒸H2O 补足10 μL。循环程序: 94℃15 min, [94℃30 s,60℃1 min, 72℃30 s] 20 个循环。为了探索最佳的退火温度,设计了温度梯度实验(56℃, 58℃, 60℃, 62℃, 64℃),电泳结果表明,在上述温度范围内PCR 效率没有明显的差异,因此所有引物的理论退火温度均为60℃,故选用60℃为统一的退火温度。第二轮添加条形码的PCR 反应体系为10 μL,以3 μL 第一轮PCR 产物为模板,1 μL1×10 PCR 缓冲液(含15 mmol/μL Mg2+),1 μL1×PCR 辅助剂,1 μL 200 μmol/μL dNTPs, 0.1 μL 条形码引物(0.2 μmol/μL),0.6 μL 25 mmol μL Mg2+,0.1 μL 热启动DNA 聚合酶(5 U/μL),再加重蒸H2O 补足10 μL。循环程序:94 ℃15 min, [94 ℃30 s,60 ℃ 1 min,72 ℃30 s]20 个循环。第二轮产物经磁珠纯化后,作为建库模板,10 μL 的反应体系,以3 μL 第二轮PCR 产物为模板,1 μL1×10PCR 缓冲液(含15 mmol/μL Mg2+),1 μL 1×PCR 辅助剂,1 μL 200 μmol/μL dNTPs, 0.1 μL P5 与P7 引物(0.5 μmol/μL), 0.6 μL 25 mmol/μL Mg2+, 0.1 μL 热启动DNA聚合酶(5 U/μL),再加重蒸H2O补足10 μL。PCR程序为: 94℃ 15 min,[94℃ 30 s, 60℃90 s,72℃30 s]15 个循环,72℃10 min[5]。建库产物送金唯智生物(苏州金唯智生物科技有限公司, 中国苏州)进行高通量测序, 使用机型为illumina X-10, 上机前产物经安捷伦2100质控。
1.7 测序数据分析和SNP 鉴定
首先利用FASTQC[6]对原始序列进行质控,质控完通过使用FASTX-Toolkit[7],根据条形码序列的错配碱基参数小于1,分离出所有样本。随后,使用Cutadapt[8]软件切除全部接头,获得每个样品的靶序列。通过使用BWA(v0.7.12)[9]和Samtools(v0.1.19)[10]的软件鉴定SNP 位点。简言之,将靶序列通过BWA比对到SNP参考序列(小鼠参考基因组mm10),使用Samtools,将sam文件生成mpileup文件,该文件用于SNP位点碱基的统计。对于SNP的检出,过滤小于15×测序深度位点,杂合子判定标准为等位基因的序列读长比例在20%~80%。
2 结果
2.1 扩增子均一性
在某批次小鼠样本测序中,6 个样本的原始数据总量为685 M,干净(clean)数据为568 M,平均深度为5308×。在这批样本中总扩增子数量为714 个,有效扩增子的数量为97%,扩增子测序深度的中值为3175×。根据SNP 鉴定时,有效深度不低于15 ×,本批样本的有效扩增子数量为97%,即97%的扩增子最后获得SNP 数据(图3)。
随后, 每个扩增子深度对平均深度进行了归一化,如此则可直接观察到平均测序深度对每个扩增子的影响, 即可评价总体均一度。从图4 可以看出,约80%的数据分布于平均深度的5倍范围以内,较高的总体均一度,使得总体测序量得以降低。
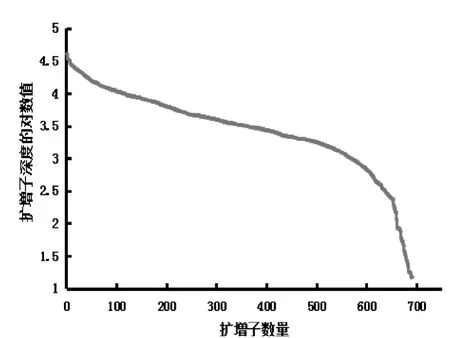
图3 扩增子测序深度Figure 3 Amplicon sequencing depth

图4 扩增子相对深度累积曲线Figue 4 Normalized coverage distribution plots
2.2 Hi-SNP 特异性
从各SNP 等位基因所在序列读长比例(图5)看,该批次全部样本为纯合子(<20%或>80%),而该批次样本均为核心群近交系小鼠,符合遗传质量检测的要求。
同时,我们对该批次不同品系小鼠分别进行靶向Hi-SNP 高通量重测序与LDR 鉴定。如表1 所示,Hi-SNP 结果与LDR 结果完全吻合。
2.3 遗传质量检测的有效性
检测了4批小鼠样本(共98个),结果表明不同来源的同一品系SNP 状态完全相同。在4 批样品中,共选择了112 个SNP 位点,SNP 位点出的比例分别为99.82%,92.00%,99.10%和90.35%(表2)。同时,样品在这些位点处基因型是纯合的,这证实这些小鼠品系都是纯系。
如表2 中所示,4 次测序获得总数据量为32 M序列读长,经过滤后得到干净数据为9 M。最低平均深度为212×,最高深度为9542×,位点鉴定成功率大于90%,样本鉴定成功率为100%。
2.4 SNP 位点在品系间的的差异分析
SNP 位点在品系间的差异数量决定了分辨率。针对常见的10 个品系,在两两品系之间进行位点差异比较,最大差异数为73 个, 最小差异数为3个,差异位点平均数为53个, 差异中位数为60个(图6)。
3 讨论
多重PCR 靶向二代测序SNP 分型方法相比于形态学、免疫学以及生物化学方法有着明显优势,主要体现在通量大、建库方便、测序深度高、性价比高、特异性强、分辨率高和价格低廉等方面。

图5 SNP 位点等位基因序列读长比例Figure 5 SNP sites allele reads ratio

表1 Hi-SNP 法与LDR 结果对应表(部分)Table 1 Table of Hi-SNP method and LDR results (partial)

表2 数据质控结果以及SNP 位点分析结果Table 2 Data quality control results and SNP sites analysis results

图6 任意两个品系差异等位基因数目的分布图 Figure 6 Distribution of the number of alleles in any two strains
多重PCR 靶向二代测序方案,在两端添加的条形码可以一次对上万个样本进行标记,并在单次上样可得全部序列,使得高通量的样本得以快速有效鉴定。在建库过程中, 只需几步PCR 反应扩增,纯化后可以直接进行测序,建库方便。在费用上,考虑到PCR 的试剂费用以及一部分不当损耗,分摊到每个样本, 引物加上反应试剂的费用微不足道。同时, 测序一个流通槽的费用大约7 000~8 000 元,一个流通槽的大小约为90 G, 全基因组测序深度为10~30X,所需数据量为30~90 G,而靶向二代测序SNP分型方案的测序深度达到了数百X甚至数千X(表2),100 个样本所需数据量为4.5 G,90 G可产生2 000 只小鼠的数据。分摊到每只小鼠的测序费用极低。
等位基因比例与PCR-LDR 方法验证表明,靶向二代测序SNP 分型方案的特异性很高。对于纯合子个体样本,在统计等位基因数量时,应只出现一种等位基因(图5),等位基因所在的序列读长比例趋近于0 或1,全部的样本都为纯合子。同时部分小鼠的结果与PCR-LDR 完全一致,可见所采集的近交系小鼠符合遗传质量检测要求。
本研究建立的靶向二代测序SNP 分型方案,相比于PCR-LDR 分型方案[11],分辨率大大提高。同时, 112个SNP位点对小鼠进行遗传监测的方案比Harlan 实验室采用48 个位点的SNP 库对小鼠进行遗传检测的方案更加有效,通过这112 个SNP位点的信息完全可以鉴定近交系小鼠的品系。
就遗传质量控制的DNA 技术方法而言,对中国知网文献调研可知, 从1986年至今涉及小鼠40篇以上,研究深度上,从早期的血清学研究[12]、到1990年代的DNA指纹[13]与延续至今的DNA微卫星技术[14], 及至最近的SNP 鉴定方案[15,16]。从理论上讲, 品系间多态性越好的位点越适合进行品系鉴定,但从有关近交系小鼠最初的培育奠基者数量极少的现实与检测结果来看,短串联重复(STR)往往在不同品系间条带一致, 仅个别位点能进行区分[17]。
鉴于国内应用SNP 标记分析通量相对较低,尚未建立针对我国常用小鼠进行系统而有效遗传检测的高通量SNP 位点组合(SNP panel),SNP 遗传检测的方法及判定标准上的现状,本研究通过高通量多重PCR 技术联合二代测序,优化出一套可用于小鼠品系遗传质量快速检测的高通量SNP 鉴定方法,易于标准化流程,非常有利于提高我国的小鼠遗传质量控制的标准。
猜你喜欢
区域治理(2022年40期)2022-11-27
中老年保健(2022年1期)2022-08-17
保健医苑(2022年5期)2022-06-10
今日农业(2020年14期)2020-12-14
动漫界·幼教365(小班)(2019年10期)2019-10-28
动漫界·幼教365(大班)(2019年10期)2019-10-28
动漫界·幼教365(中班)(2019年10期)2019-10-28
中华建设(2019年7期)2019-08-27
山东林业科技(2019年2期)2019-06-03
云南农业科技(2019年2期)2019-04-04
