
二叶式主动脉瓣钙化发病机制及相关蛋白的研究进展
2019-05-16 02:01:00
国际心血管病杂志 2019年2期
二叶式主动脉瓣(BAV)是人类常见的先天性心脏病,发病率为0.5%~2%,其中20%~50%的患者因发展为重度主动脉瓣狭窄而行主动脉瓣置换术[1]。BAV导致的BAV钙化(BAVC)通常合并主动脉病变,晚期易发生升主动脉瘤或主动脉夹层等危及生命的并发症[2-3]。过去认为主动脉瓣钙化是一种退行性病变,由钙盐在瓣膜表面被动沉积导致。然而,也有研究表明,主动脉瓣钙化是一个活跃的病理生理过程,具有较强的遗传特性,其机制与成骨机制相似[4-5]。影响瓣膜钙化的因素多种多样,包括氧化应激、细胞增生、成骨作用、细胞炎性反应、细胞凋亡和坏死等[6]。BAVC的发病机制目前尚未明确。
正常主动脉瓣包括纤维层(fibrosa)、松质层(spongiosa)、心室基层(ventricularis)。瓣膜内皮细胞(VEC)位于瓣膜血液接触面,调节瓣膜的通透性、炎性细胞的黏附和旁分泌信号。主动脉瓣间质细胞(AVIC)分布于主动脉瓣各瓣膜层,是瓣膜主要的细胞类型。AVIC是瓣膜重构的关键,调节细胞外基质(ECM)的合成和降解[7]。从瓣膜形态上看,钙化瓣叶纤维层增厚,纤维膜附近钙化结节形成[8](见图1)。电镜下可见瓣膜钙化组织与骨组织相似,其主要成分为羟基磷灰石结晶[9],其钙化程度可通过冯科萨染色判断(见图2)。
1 BAVC相关蛋白
1.1 骨形态发生蛋白(BMP)
BMP-2、BMP-4是主动脉瓣钙化的重要调节因子。BMP-2的下游因子如runt相关转录因子2(RunX2),与α-平滑肌肌动蛋白(α-SMA)一起作为肌成纤维细胞和成骨细胞的分化标记物,参与调控成纤维细胞分化和骨形成[6,10]。Gomez-Stallons等[11]在对培养了6周的Klotho-/-基因敲除小鼠的研究中发现,实验组主动脉瓣钙化结节区有BMP-2高表达,而无明显钙化的对照组中主动脉瓣未见明显BMP-2表达。此外,Klotho-/-实验组中BMP-2、BMP-4配体基因表达也较对照组增加。Zeng等[12]通过抑制白细胞介素-37的表达,发现人AVIC钙化沉积明显加重,同时BMP-2和碱性磷酸酶(ALP)表达增加。Sun等[6]在研究剪切应力对瓣膜钙化影响的实验中发现,剪切应力增加的BAV实验组较对照组BMP-4水平增加2.4倍。Yanagawa等[13]发现BAV钙化组BMP-2及α-SMA的表达水平较对照组显著升高,并且RunX2表达水平也相对升高,这进一步证明了BMP-2和BMP-4与BAVC相关。
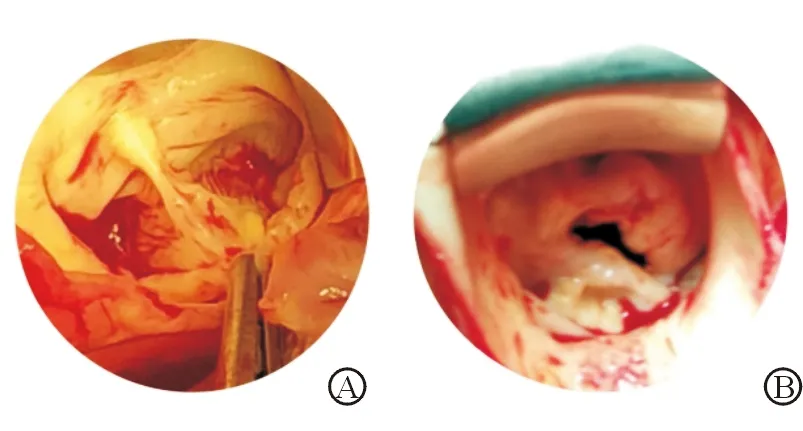
注:A为非钙化组织;B为钙化组织;图片来自中南大学湘雅二医院心脏中心
图1 二叶式主动脉瓣
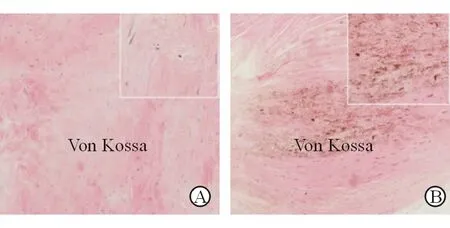
注:A为非钙化组织;B为钙化组织;图片来自中南大学湘雅二医院心脏中心
图2 主动脉瓣组织的冯科萨染色
1.2 基质金属蛋白酶(MMP)
MMP是一种锌离子依赖性明胶酶,在ECM降解及重构过程中有重要作用,并受其活化程度、基因表达水平以及金属蛋白酶组织抑制剂(TIMP)共同调节[14]。有研究表明,MMP如MMP-2、MMP-9、MMP-12的升高与主动脉瓣钙化有关。Sun等[6]研究显示,剪切应力增加的BAV实验组中MMP-2和MMP-9的表达水平较对照组明显升高。Jung等[15]给4~5周的ApoE-/-基因敲除小鼠分别喂饲高脂饮食3、6、9个月后,小鼠主动脉瓣钙化发生率分别为0%、17%、50%;通过RP805示踪剂特异性激活MMP,并应用显微SPECT及增强CT间接测量MMP活化水平,结果发现MMP活化及MMP-12表达水平6个月组>9个月组>3个月组,这提示MMP活化水平可能与主动脉瓣钙化有关,但并不呈线性正相关发展。Du等[10]通过冯科萨染色选取钙化沉积相当的BAV、风湿性三叶主动脉瓣(RTV)及退行性变三叶主动脉瓣(DTV),免疫组织化学染色显示BAV组中MMP-2和MMP-9的蛋白表达水平较RTV、DTV组显著升高,天狼星红染色显示BAV较RTV和DTV组织有更多的胶原蛋白表达。由此说明MMP在主动脉瓣钙化过程中起重要作用,且在BAVC中更为突出。
1.3 Smad蛋白家族
Smad蛋白家族是一类细胞内信号传导蛋白。多项研究表明,Smad蛋白磷酸化与瓣膜钙化相关,并能直接参与转化生长因子-β(TGF-β)超家族成员的信号传导[10-11,16-17]。Ankeny等[16]研究发现,人类主动脉瓣钙化区内出现了磷酸化Smad1/5/8 (pSmad1/5/8)的高表达,表明pSmad1/5/8水平与主动脉瓣钙化有关。Du等[10]研究发现,BAV与RTV、DTV相比,微小RNA(miRNA)-195表达水平降低,而Smad7表达水平升高;并且miR-195可以直接作用于Smad7,进而调节瓣膜钙化水平。这提示Smad蛋白可能对BAVC有促进作用。
1.4 Sox9蛋白
Sox9是软骨形成的主要调节因子,参与软骨细胞增殖、分化和成熟过程[18]。Peacock等[19]研究发现,Sox9缺陷小鼠可出现心脏瓣膜钙化,而体外敲除Sox9可导致RunX2表达增加和钙沉积,说明Sox9有抑制瓣膜钙化的作用。但Li等[18]用高浓度磷酸盐体外培养人AVIC,发现RunX2和ALP表达增加,AVIC长时间接触磷酸盐会导致钙沉积,敲除Sox9可减弱人AVIC中磷酸盐诱导的高钙沉积,并促使RunX2和ALP表达下调。Sox9在小鼠及人瓣膜钙化的进展中作用不一,可能与不同种属间钙化机制存在差异有关。
1.5 羧基谷氨酸蛋白(MGP)
MGP是一种维生素K依赖蛋白,以两种不同的形式存在:非羧化活性(ucMGP)和羧化活性(cMGP)。与所有维生素K依赖蛋白一样,MGP需要维生素K诱导其羧基化来发挥作用。MGP主要通过抑制BMP表达以及与羟基磷灰石直接作用这两条途径来阻止钙化进程[20]。Chiyoya等[21]使用30 ng/mL肿瘤坏死因子-α (TNF-α)体外诱导人AVIC,发现实验组细胞钙化程度加重,并出现MGP低表达和BMP表达上调,进一步敲除MGP基因可以显著诱导钙化,并引起BMP基因表达升高。由此说明MGP可以通过特异性抑制BMP表达,延缓主动脉瓣膜钙化进程,但其是否可以同样抑制BAVC中BMP表达,需进一步验证。
1.6 一氧化氮合酶(NOS)
一氧化氮(NO)由内皮细胞分泌,在许多生理和病理过程中起着关键作用,并与主动脉瓣钙化过程有关[3]。Kim等[22]研究发现,与正常三叶主动脉瓣膜(TAV)组相比,BAV组75%的患者存在内皮源性NOS(eNOS)、TIMP-2表达下调,而MMP-9表达上调。El Accaoui等[23]在对eNOS-/-小鼠的研究中发现,eNOS表达下降更容易导致BAVC,进而证实NOS与BAVC相关。
1.7 成骨生物标志蛋白
骨桥蛋白(OPN)、ALP以及RunX2是成骨细胞的标志物,大量研究证明其与瓣膜钙化相关,并作为瓣膜早期钙化的标志物,在BAVC中高表达[12,17,24-26]。Song等[17]发现,抑制TGF-β1和BMP-2活性可降低ALP、OPN、RunX2的表达水平,抑制钙沉积,提示ALP、OPN、RunX2与瓣膜钙化相关,并受TGF-β1和BMP-2调控。
2 蛋白相关信号通路
2.1 Notch信号通路
Notch1信号通路是参与细胞生长、细胞分化和心脏瓣膜形成的高度保守信号通路。在常染色体显性遗传家系中,Notch1突变是引起BAV的重要原因,Garg等[3]研究认为,Notch信号通路的丢失可通过RunX2、Sox9和BMP-2依赖机制导致主动脉瓣钙化。Nus等[24]使用γ分泌酶抑制剂DAPT抑制猪AVIC的Notch信号表达,通过茜素红染色发现DAPT组钙化结节较空白对照组增加2倍,同时BMP-2、RunX2、ALP均表达上升。Irtyuga等[5]研究发现,钙化狭窄的BAV/TAV患者血清中骨保护蛋白(OPG)显著高于对照组,且可溶性核因子κB受体激动剂配体(sRANKL)仅在BAV组中明显升高。此外,Notch1突变组OPG表达上调,OPG/RANKL比值也明显升高,由此推测Notch1可能通过OPG/RANKL/RANK信号通路促进瓣膜钙化。也有研究结果与之相反,Zeng等[25]在对人AVIC的研究中发现,加入NICD1抑制剂DAPT的实验组BMP-2、ALP的表达水平较对照组降低,更重要的是细胞内钙盐沉积程度减轻。Kostina等[26]从人BAVC、钙化TAV以及非钙化TAV中分离培养AVIC,发现BAVC患者的AVIC对早期成骨分化刺激的敏感性明显高于其他两组,对Notch信号引起的OPN、ALP表达活化也更为敏感。Notch在不同种属、不同个体中作用各异,可能与其复杂的遗传信息相关,因此Notch在BAVC中的具体分子机制有待进一步研究。
2.2 TGF-β超家族信号通路
2.2.1 BMP信号通路 BMP-2是目前研究最多、诱导成骨分化及促进骨形成作用最强的细胞外信号分子之一,其主要通过BMP-2/RunX2信号通路参与成骨分化,BMP-2与其受体结合后可以激活细胞外信号调节激酶1/2 (ERK1/2)或Smads复合物,正向调节RunX2,再激活下游osterix基因,促进骨形成及成骨分化。Yanagawa等[13]认为AVIC的上游活化剂如TGF-β可以触发BMP-2进而激活ERK1/2,从而正向调节RunX2,导致钙化和主动脉瓣狭窄。Song等[17]使用双糖链蛋白多糖诱导AVIC后,发现ALP、OPN和RunX2的表达较对照组显著增加,BMP-2拮抗剂降低了其诱导的ALP、OPN和RunX2的蛋白水平,由此证明BMP-2在主动脉瓣钙化中起关键作用。进一步敲除Smad1基因,发现双糖链蛋白多糖诱导的ALP、骨桥蛋白和RunX2的蛋白水平较前下降,提示BMP-2可以通过激活Smad1正向调节RunX2,从而促进骨形成及成骨分化,导致瓣膜钙化。
2.2.2 TGF-β信号通路 研究证明,TGF-β1和TGF-β3与主动脉瓣钙化相关,但其作用有所不同[10,27-28]。Miller等[27]在AVIC体外培养实验中发现,TGF-β1可以诱导细胞凋亡、细胞聚集和钙化结节形成。Du等[10]通过10 ng/mL TGF-β体外诱导AVIC,发现TGF-β组钙化水平较对照组提高3.9倍。Song等[17]在双糖链蛋白多糖诱导的AVIC实验中也证实了TGF-β1在主动脉瓣钙化中的关键作用,进一步敲除Smad3基因,其诱导的ALP、OPN和RunX2的表达水平较前下降,表明TGF-β1可以通过激活Smad3正向调节RunX2,从而促进骨形成及成骨分化,导致瓣膜钙化。Fang等[28]则在AVIC实验中发现,抑制miR-29b基因可上调TGF-β3及下调Wnt3/β-catenin/Smad3的表达,由此证明miR-29b可通过抑制靶标TGF-β3的表达导致瓣膜钙化。
2.2.3 活化素蛋白激酶2(ALK2)信号通路 ALK2属于TGF-β超级家族的Ⅰ型受体成员之一。Thomas等[29]在对ALK2基因敲除小鼠的研究中发现,ALK2基因缺失更容易导致BAV及主动脉瓣狭窄,虽未使瓣膜出现明显钙化,但可诱导细胞增殖,同时可导致ERK1/2磷酸化水平增加,促进成骨因子的表达,由此说明ALK2可能在BAVC过程中起作用。
2.2.4 骨形态发成蛋白受体IA(BMPR-IA)通路 BMPR-IA是TGF-β超家族成员之一。研究显示,BMPR-IA与瓣膜钙化相关。Gomez-Stallons等[11]在Klotho-/-小鼠中发现,BMPR-IA失活可以导致主动脉瓣钙化减轻,pSmad1/5/8表达下降,提示在Klotho-/-小鼠中BMP-pSmad1/5/8信号通路对主动脉瓣钙化起重要作用。此外,在猪AVIC的体外实验中发现,LDN-193189(BMPR-IA抑制剂)处理可以显著抑制pSmads1/5/8表达,这进一步表明BMPR-IA通过pSmad1/5/8调节下游成骨因子,导致瓣膜钙化。
2.3 ERK1/2信号通路
只有磷酸化的ERK1/2才具有活性并与瓣膜钙化有关。Zeng等[25]在人类的AVIC体外试验中发现,脂多糖实验组可以激活ERK1/2磷酸化并高表达BMP-2、ALP。而加入ERK1/2抑制剂PD98059可以降低脂多糖引起的ERK1/2磷酸化水平及BMP-2、ALP表达升高,提示ERK1/2在主动脉瓣钙化过程中起重要调控作用。
2.4 核因子κB(NF-κB)信号通路
Zeng等[25]在对人AVIC的研究中发现,脂多糖实验组与空白对照组相比,可以诱导BMP-2、ALP高表达;而NF-κB抑制剂SN50可以降低脂多糖引起的BMP-2、ALP的表达,提示NF-κB在主动脉瓣钙化过程中起重要调控作用。Patel等[30]用14%循环拉伸力模拟BAV环境,体外培养AVIC,转染miR-148a-3p组白细胞介素和MMP的表达水平较对照组降低,NF-κB抑制蛋白(IκB)表达增加,提示miR-148a-3p可以抑制NF-κB信号通路,引起白细胞介素和MMP表达降低,推断NF-κB信号对BAVC有促进作用。
2.5 Wnt信号通路
Wnt信号通路普遍存在,功能广泛,而其中最为经典的为Wnt/β-catenin信号通路。Carrion等[31]用Wnt激动剂体外培养AVIC 48 h,发现实验组ALP和BMP-2的mRNA水平较对照组明显升高;同时,拉伸应力组β-catenin mRNA表达水平及其靶基因CCND1表达水平较对照组分别增加39%、91%,且拉伸应力可以激活AVIC中的Wnt/β-catenin信号通路,提高钙化相关基因的表达。这进一步证实Wnt/β-catenin信号可能通过调控ALP和BMP-2水平促进BAVC发展。
3 信号通路的相互关系
过去研究认为各个信号通路是独立发挥作用的,但现在越来越多的学者认为各信号通路存在相互影响、彼此交叉的关系(见图3)。Notch通路、TGF-β通路、NF-κB通路、Wnt通路均可通过上调BMP-2水平促进pSmad1/5/8表达,进而正向调控成骨相关因子ALP、OPN、RunX2的表达,导致成骨分化及瓣膜钙化。同时,Notch通路对NF-κB通路亦有调控作用,并可直接激活ERK1/2磷酸化,从而调控下游成骨因子的表达。
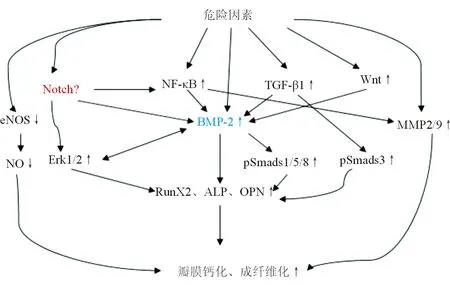
图3 信号通路关系图
4 总结及展望
BAVC的发生和发展是多因素、多通路综合作用及交叉影响的结果。目前对其完整的发病机制尚无明确统一的论述。通过对BAVC相关蛋白进行研究,有望进一步了解BAVC病因,利用BAVC相关蛋白检测筛选高危人群,针对蛋白靶点进行干预治疗,延缓BAVC进程。
猜你喜欢
口腔医学(2021年10期)2021-12-02 02:08:00
心肺血管病杂志(2020年5期)2021-01-14 00:43:30
心肺血管病杂志(2019年6期)2019-07-12 09:04:34
心肺血管病杂志(2019年4期)2019-06-27 07:36:10
心肺血管病杂志(2019年1期)2019-04-22 01:12:04
大学生(2017年10期)2017-10-23 18:35:06
中华老年口腔医学杂志(2016年2期)2017-01-15 14:24:47
中国病理生理杂志(2015年8期)2015-12-21 12:38:14
天津护理(2015年4期)2015-11-10 06:11:41
天津医科大学学报(2015年3期)2015-06-05 12:21:49
