
基于线粒体控制区的中国近海棘头梅童鱼群体遗传结构研究
2019-05-10 07:04梁述章马春艳蒋科技
海洋渔业 2019年2期
梁述章,宋 炜,马春艳,蒋科技,
张凤英1,赵 明1,马凌波1
(1.中国水产科学研究院东海水产研究所,农业部远洋与极地渔业创新重点实验室,上海 200090;2.上海海洋大学水产与生命学院,上海 201306)
棘头梅童鱼(Collichthys lucidus),属鲈形目(Perciformes),石首鱼科(Sciaenidae),梅童鱼属,为短距离洄游的浅海鱼类,喜栖息在河口咸淡水交汇处,生长速度快,适温、适盐范围广[1-4]。其肉质细嫩可口,沿海居民喜食此鱼,而目前市场销售的棘头梅童鱼数量有限,价格居高,无法满足市场需求[5]。近几年中国水产科学研究院东海水产研究所研究团队加大了棘头梅童鱼人工繁育和驯养技术的攻关力度,取得了可喜的成果。而对棘头梅童鱼遗传结构的认识,也是该物种保护和合理利用的重要内容。
鱼类线粒体DNA(mt DNA)与其它许多脊椎动物的mt DNA一样,结构简单,母系遗传,几乎不发生重组,进化速度快,是鱼类分子群体遗传学和分子系统学理想的分子标记[6-8]。控制区(D-loop区)是mt DNA上的一段非编码区,在mt DNA上,D-loop区的进化速度最快,较适合用于种群遗传结构分析和系统发育分析[9-10]。目前,仅见郑德峰等[11]和殷丽娜[12]利用线粒体控制区对棘头梅童鱼的遗传特征和遗传分化进行了初步分析,为更好地认识我国沿海棘头梅童鱼群体遗传结构现状,本研究利用线粒体D-loop区对棘头梅童鱼7个不同地理群体的遗传多样性进行评价,以期为棘头梅童鱼资源的保护和利用提供基础材料。
1 材料与方法
1.1 实验材料
实验用鱼分别采集于江苏连云港、江苏大丰、上海崇明、浙江舟山、浙江温州、福建宁德和福建厦门海域(图1)。样品采集地点、时间、数量和体长等信息见表1。
1.2 DNA提取、扩增及测序
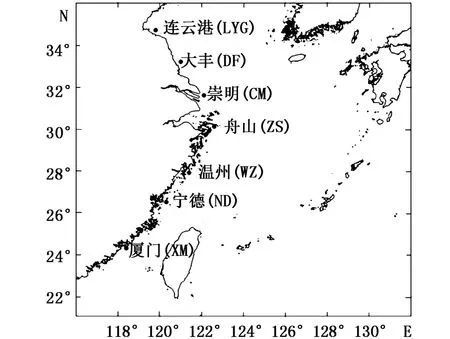
图1 样本采集地点Fig.1 Map of the sampling locations注:LYG:Lianyungang;DF:Dafeng;CM:Chongming;ZS:Zhoushan;WZ:Wenzhou;ND:Ningde;XM:XiamenNote:LYG:Lianyungang;DF:Dafeng;CM:Chongming;ZS:Zhoushan;WZ:Wenzhou;ND:Ningde;XM:Xiamen
样本冰冻保存运输至实验室进行肌肉组织取样,用于DNA提取。采用海洋动物组织基因组DNA提取试剂盒(北京,天根生化科技),以1%的琼脂糖凝胶电泳检测DNA质量,分光光度计检测其浓度和纯度。线粒体D-loop区域的引物序列为 AAATCCTTGAATAACACCGC(DL-F)和 ATCACTGCTGAGTTCCCTTG(DL-R),由上海杰李生物技术有限公司合成。PCR反应体系为25μL:模板 DNA 1μL、酵母 PCR Mix 12.5μL、上下游引物各1μL,加双蒸水至总体积25μL。PCR反应条件为:94℃预变性3 min,94℃变性25 s,58℃退火50 s,72℃延伸90 s,共40个循环;72℃最后延伸10 min;4℃保存。扩增产物经1.2%琼脂糖凝胶电泳检测后送至上海杰李生物技术有限公司正反双向测序。
1.3 数据分析
序列经拼接后,于 NCBI数据库中进行BLAST同源检测,利用Clustal X软件对序列进行比对排序与校正;MEGA 5.1软件用以统计群体的突变位点数、突变类型及核苷酸组成,计算群体内和群体间的遗传距离[13];用 DnaSP 5.1软件确定序列的单倍型数、单倍型多样性、核苷酸多样性以及平均核苷酸差异数等参数[14];基于Kimura双参数模型构建群体间单倍型的NJ树,可靠性经 1000次重复抽样检验[15-16];应用Arlequin 3.1软件的分子方差分析(AMOVA)进行群体间的遗传变异评估,通过1000次重抽样来检查不同遗传结构水平上协方差的显著性[17]。采用分化固定指数Fst来评价两两群体间的遗传差异,通过1000次重抽样来检查两两群体间的Fst显著性,通过 Fst值计算基因流 Nm[18]。采用Network软件构建单倍型网络图,检测单倍型间的进化关系,并进行中性检验和错配分析,推测种群的平衡状态以及种群事件时间[19]。
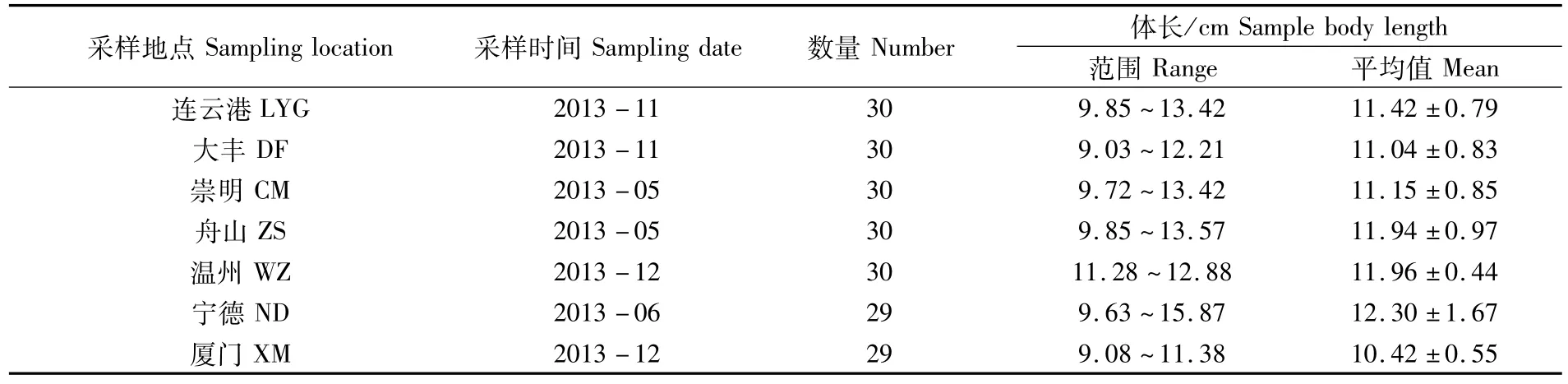
表1 棘头梅童鱼采样信息Tab.1 Sampling details of C.lucidus
2 结果与分析
2.1 D-loop区序列分析
PCR扩增产物经测序及序列比对得到795 bp的D-loop基因序列,7个群体的线粒体控制区碱基组成基本一致,A、T、C、G平均含量分别为31.04%、31.43%、23.99%和 13.54%,A+T含量(62.47%)明显高于 G+C含量(37.53%),与大多数脊椎动物线粒体DNA的碱基组成相似,体现出显著的AT碱基偏好性[20]。208条序列共检测到66个变异位点,存在4个插入/缺失现象,转换颠换比 K嘌呤=4.99,K嘧啶=7.813,整体 R=2.845。
2.2 遗传多样性分析
7个群体共检测到83种单倍型,未发现所有群体的共享单倍型,每个群体均有独享单倍型,其中CM群体最多,有17个独享单倍型(表2)。CM、LYG、DF和 ZS群体之间存在共享单倍型Hap1,占所有个体的22.12%,WZ、ND和 XM群体之间存在两个优势单倍型Hap45和Hap47,共占整体的23.08%。从单倍型分布可以看出7个群体之间存在地理上的分化现象。
群体的遗传多样性参数见表3。单倍型参数方面,CM和WZ群体的单倍型数目及单倍型多样性较高,分别为20(0.954 00±0.000 67)和19(0.948 00±0.000 77),XM群体的单倍型数目和单倍型多样性最低,仅为 7(0.554 00±0.009 98)。核苷酸参数方面,DF群体的核苷酸多样性和平均核苷酸差异数最高,分别为0.007 08和5.621,XM群体最低,分别为0.001 83和1.448。由以上结果可见,CM、WZ和DF群体遗传多样性高,而XM群体的遗传多样性最低。

表2 棘头梅童鱼7个群体单倍型分布情况Tab.2 Distribution of haplotypes among 7 populations of Collichthys lucidus

·续表·

·续表·
2.3 群体遗传结构
棘头梅童鱼不同群体内遗传距离在0.002~0.007之间,其中DF群体的内部遗传距离最大,XM群体内部遗传距离最小(表4)。两两群体间的遗传距离在0.004~0.039的范围内,LYG、DF、CM和ZS 4个群体之间的遗传距离及WZ、ND和XM 3个群体之间的遗传距离较小,在0.004~0.007之间,而两组群体间的遗传距离在0.036~0.039之间,说明7个棘头梅童鱼群体之间明显分为两支,即北方群体(LYG、DF、CM和ZS)和南方群体(WZ、ND和XM)。
7个棘头梅童鱼群体间遗传分化结果如表4所示,两两群体的遗传分化系数 Fst值在-0.008 72~0.928 28之间。DF和 LYG群体的Fst值为 -0.008 72,遗传分化程度极低;CM、ZS群体和DF、LYG群体两两之间的Fst值属于中度遗传分化;ND、WZ和XM群体之间的Fst值均在0.05之内,属于低度遗传分化;DF、LYG、CM和ZS群体与其它3个群体间的Fst值均大于0.8,说明两组地理群体间遗传结构存在显著的差异。
基于Fst值计算出群体间的基因流Nm,LYG、DF、CM和ZS群体两两之间的Nm值均大于1,与其它3个群体的Nm值极小;WZ、ND和XM群体两两之间的结果一致(表5)。
上述结果作为依据,将7个棘头梅童鱼群体分为2组,进行遗传差异的分子方差分析。总的方差分为组间方差组分(Va)、组内群体间方差组分(Vb)和群体内方差组分(Vc),如表6所示,组间变异为85.97%,组内群体间变异占0.68%,群体内变异为 13.35%,遗传分化指数 Fst=0.866 47(P=0)。
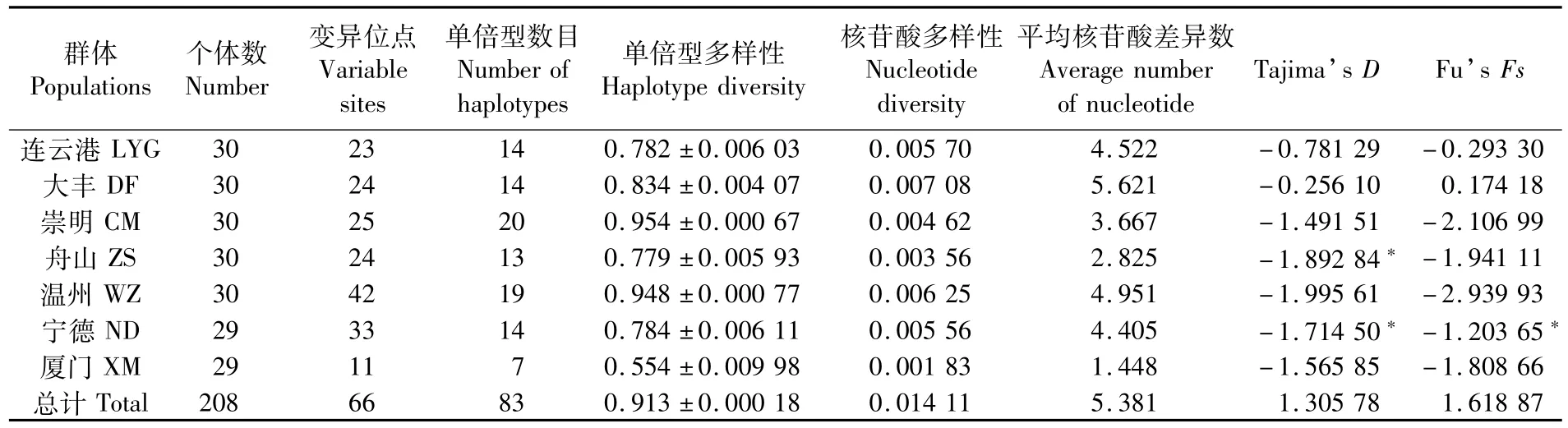
表3 棘头梅童鱼D-loop区遗传多样性参数Tab.3 Genetic diversity parameters of mt DNA D-loop region in Collichthys lucidus

表4 棘头梅童鱼群体内遗传距离(对角线,粗体)及两两群体间遗传距离(对角线上)和遗传分化系数(F st)(对角线下)Tab.4 Pairwise genetic distances within population(diagonal,bold),and genetic distance(above diagonal),fixation index(F st)(below diagonal)between every two populations of Collichthys lucidus
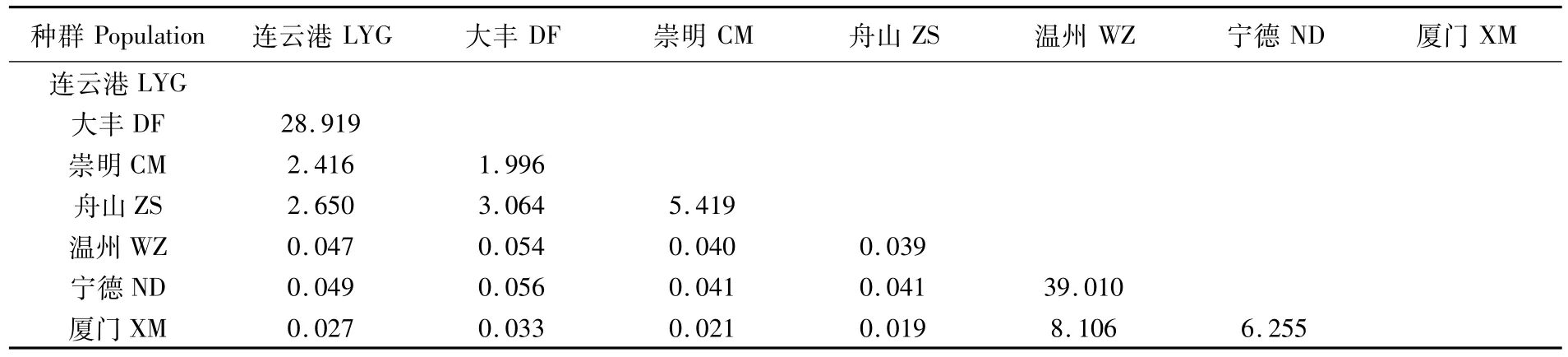
表5 棘头梅童鱼群体间的基因流Tab.5 Gene flow value between populations of Collichthys lucidus

表6 棘头梅童鱼群体间遗传差异分子方差分析Tab.6 Analysis of molecular variance(AMOVA)of populations of Collichthys lucidus
2.4 单倍型邻接关系树和简约网络图
为了更直观地阐述群体间的遗传结构,以中介邻接网络法构建了棘头梅童鱼单倍型网络图(图2),单倍型被分为3部分,其中两部分分别以Hap1和Hap47为中心呈典型的星状分布态势逐级拓展,另一部分作为过渡单倍型存在。Hap1存在于LYG、DF、CM和 ZS群体中,Hap47存在于WZ、ND和XM群体中。
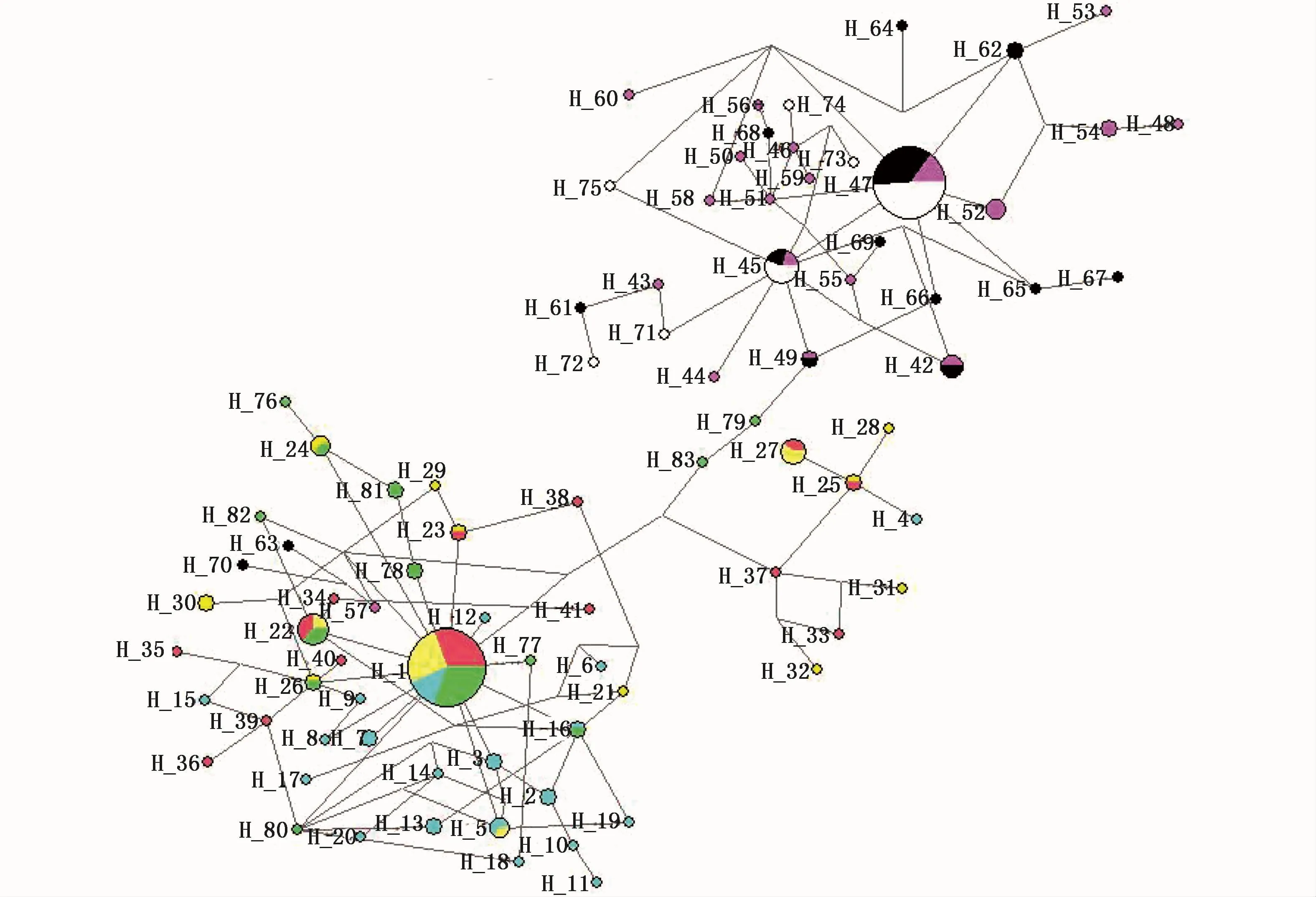
图2 基于D-loop区构建棘头梅童鱼单倍型网络关系图Fig.2 Haplotype network diagram constructed based on D-loop region of Collichthys lucidus注:连云港:粉色;大丰:黄色;崇明:蓝色;舟山:绿色;宁德:紫色;温州:黑色;厦门:白色Note:LYG:Pink;DF:Yellow;CM:Blue;ZS:Green;ND:Purple;WZ:Black;XM:White
以单倍型构建邻接关系树,大部分节点分支的支持率均大于50%,具有显著的地理谱系结构(图3)。邻近关系树中大致分为两个区域,结合表2可知,区域1的单倍型基本出现在LYG、DF、CM和ZS群体中;区域2的单倍型基本出现在WZ、ND和XM群体中,有少数ZS群体个体出现。单倍型邻接关系树和单倍型简约网络图将7个棘头梅童鱼群体分为2个地理群体,与遗传距离分析和Fst值所得结论一致。
2.5 中性检验和错配分析
棘头梅童鱼群体中性检验包括Tajima’s D和 Fu’s Fs分析,如表3所示,Tajima’s D的检测结果均为负值,ZS和ND群体显著,其余群体不显著;Fu’s Fs检验结果中,除DF为正值外,其它均为负值,且仅ND群体显著。上述结果表明,各个群体,尤其是ND和ZS群体,符合中性模型,选择压力小,可能经历了种群的规模性扩张或定向选择。错配分析结果如表7所示,整体的SSD值为0.161 42(P>0.05),各个群体的值在 0.005 29~0.658 15之间,Rag整体值为0.040 71(P>0.05),说明棘头梅童鱼仍处于膨胀阶段,为棘头梅童鱼种群扩张提供了证据。
3 讨论
棘头梅童鱼广泛分布于中国沿海,且以黄海、东海为主。本研究选取连云港、大丰(黄海),崇明、舟山、温州、宁德和厦门(东海)7个地理群体的208个样本,利用线粒体控制区(D-loop)对棘头梅童鱼遗传多样性进行分析。
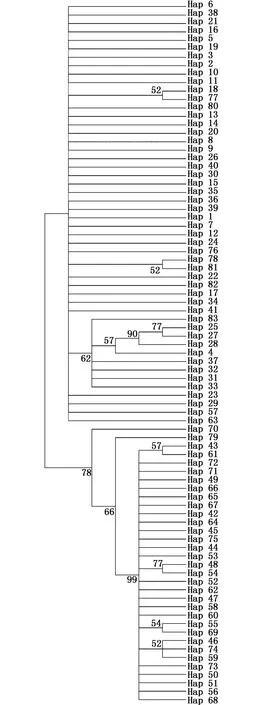
图3 基于Kimura 2-Parameter模型构建单倍型的NJ树Fig.3 NJ clustering diagram of haplotypes constructed based on Kimura 2-Parameter model
遗传多样性是生物多样性的核心,是物种生存与进化的基础,其中核苷酸多样性是分析群体遗传多样性的重要指标,与单倍型多样性共同反映线粒体DNA的变异程度[21]。本研究7个群体共发现83种单倍型,其中70种独享单倍型,分布于各个群体,无所有群体共享的优势单倍型,这与殷丽娜[12]研究结果相似,棘头梅童鱼的短距离洄游特性是造成无共享单倍型、多独享单倍型的主要原因。7个群体的总体遗传多样性高(h=0.913,π=0.014 11),且核苷酸多样性与平均核苷酸差异数呈现一致的变化趋势,说明中国沿海的棘头梅童鱼遗传资源丰富,对环境的适应能力及进化潜力大。7个群体中,XM群体的遗传多样性最低(h=0.554,π=0.001 83),根据 GRANT等[22]对海水鱼类遗传多样性划分的分布模式,判断XM群体接近第一种模式(低h低π),说明XM群体近期出现过种群瓶颈效应或种群由单一、少数系群所发生的奠基者效应形成,导致其遗传多样性偏低。其余群体均符合第二种模式(高h低π)特点,说明群体受到了环境变化的影响,经历了快速的扩张期,种群数量急剧增加,碱基的突变导致单倍型数量和单倍型多样性的增加,而核苷酸多样性没有获得足够的时间积累。Tajima’s D、Fs中性检验以及错配分析结果也证明了棘头梅童鱼经历了群体扩张,根据扩张参数τ和(3%~10%)/百万年的D-loop区进化速率推算[12],中国沿海棘头梅童鱼的群体扩张发生在3.87~12.9万年前。
遗传距离是衡量群体间和群体内部遗传关系的重要指标,遗传距离的大小反映群体间亲缘关系的远近和群体内差异程度[23-24]。本研究中群体内部的遗传距离在0.002~0.007之间,数值高低与核苷酸多样性参数的变化趋势一致。遗传距离数值将7个群体以舟山为界,明显的分为北方群体(LYG、DF、CM、ZS)和南方群体(WZ、ND、XM),两组地理群体间遗传差异显著。北方群体中,除DF群体内部遗传距离大于其与ZS、CM和LYG群体的遗传距离外,群体内部的遗传距离均小于或等于两两群体间的遗传距离,说明DF群体内部分化大,推测种群间出现了交叉;南方群体中,WZ和ND群体的内部遗传距离和两群体间遗传距离相等,说明两群体间也存在明显的交叉现象。遗传分化系数Fst是衡量群体间遗传分化程度的标准[25],本研究的Fst结果与遗传距离结果相呼应,均说明中国沿海棘头梅童鱼的遗传结构出现明显的地理区域性,即南北分化格局现象,DF和LYG群体、ND和WZ群体的Fst均为负值,同样验证两两群体间存在明显的交叉现象。WZ、ND和XM群体与北方群体的Fst值均大于0.82,且呈现地理位置上由北向南Fst值逐渐增加的趋势,XM群体最大,其与北方群体的Fst差值大于0.9,此结果与殷丽娜[12]得出的结果一致,表明棘头梅童鱼在南北分化的基础上,北方群体与南方群体地理距离越远,基因交流逐渐趋于0,分化程度越高。
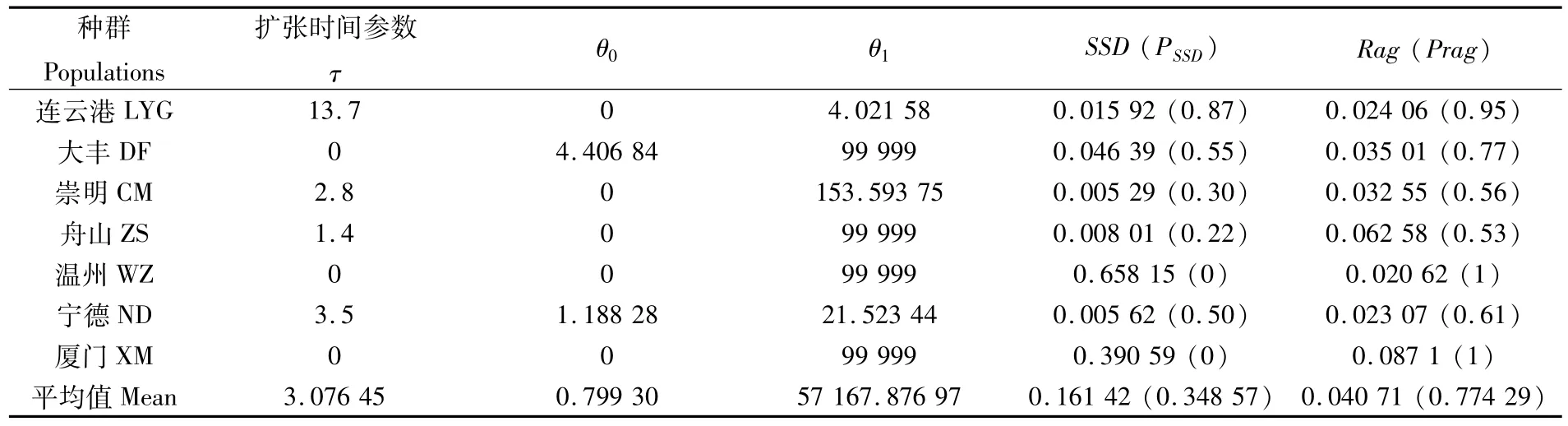
表7 棘头梅童鱼7个群体的错配分析参数Tab.7 Mismatch distribution analysis for 7 Collichthys lucidus populations
基于以上南北分化结果,计算群体的基因流Nm,并进行AMOVA分析。Nm值越大,说明群体间的基因交流水平越高[26]。本研究中,南、北地理群体间的Nm<0.1,地理群体内部Nm>2,根据WRIGHT[27]对 Nm值的划分,认为南、北地理群体之间的基因交流极弱,可能由于遗传漂变发生了分化;南、北地理群体内部存在较强的基因流,群体间的遗传分化较小。其中,DF和LYG群体、ND和WZ群体间的基因流是南北类群内部基因流的10倍以上,进一步验证遗传距离中关于其种群间交叉的结论。AMOVA分析中,组间变异为85.97%,组内群体间变异占0.68%,群体内变异为13.35%,遗传分化指数 Fst=0.866 47(P=0),说明主要的遗传变异来自于南、北两个地理群体间,群体内部也出现了一定程度的分化。基因流Nm和AMOVA分析进一步证实中国沿海棘头梅童鱼形成明显的南北分化格局。
为了更直观地表述棘头梅童鱼群体间的遗传结构,以单倍型为依据,构建邻接关系树和网络关系图。邻接关系树中,83种单倍型被分为两部分,结合单倍型分布情况可以看出,两部分分别代表南方地理群体和北方地理群体(表2)。棘头梅童鱼7个群体中存在3种优势单倍型,Hap1(北方群体)、Hap45和Hap47(南方群体),分别在南、北地理群体中适应环境变化,稳定存在。网络关系图分为主要的两部分,分别以Hap1和Hap47为中心呈星状分布态势拓展,两者结果相符。
以上结果均表明棘头梅童鱼存在明显的南北分化格局。赵明等[28]、殷丽娜等[12]和郑德峰等[29]分别利用 COI、D-loop和 AFLP分析棘头梅童鱼遗传结构,同样推断我国棘头梅童鱼存在南北分化格局;宋炜等[5]和梁述章等[30]分别利用核基因微卫星和形态框架数据分析,未发现南北分化现象。分析方法、采集样本时间、地点、数量、样本生长状况和分析方法等的不同应该是造成以上不同结论的主要原因。形态参数极易受到环境和样本生长时期的影响,线粒体分子标记由于母系遗传,易受到选择压力的影响,核基因对种群遗传结构的检测更敏感[31]。棘头梅童鱼南北分化格局产生的原因大致可归纳为以下4点:1)洋流影响:舟山是台湾暖流、日本寒流以及黄海冷流的交汇点,舟山以北主要受黄海冷流和日本寒流的影响,舟山以南受台湾暖流的影响;2)在赵明等[28]、谢起浪等[32]的研究中,均发现温州群体存在过度捕捞现象,一定程度上阻止了温州及其以南海域的棘头梅童鱼前来舟山渔场觅食、产卵等;3)棘头梅童鱼是短距离洄游种,使群体间产生基因交流,又极大程度限制基因交流的强度;4)根据扩张参数推算棘头梅童鱼群体扩张时间在3.87~12.9万年前,即更新世冰期,海平面下降导致边缘海地区的物种种群分化且产生地理隔离。
猜你喜欢
区域治理(2022年40期)2022-11-27
世界科学技术-中医药现代化(2022年3期)2022-08-22
肝博士(2022年3期)2022-06-30
清华金融评论(2022年4期)2022-04-13
国际放射医学核医学杂志(2021年10期)2021-02-28
房地产导刊(2020年7期)2020-08-24
动漫界·幼教365(小班)(2019年10期)2019-10-28
动漫界·幼教365(大班)(2019年10期)2019-10-28
动漫界·幼教365(中班)(2019年10期)2019-10-28
