
敲低lncRNA SNHG16对胃癌细胞凋亡的影响及其机制
2019-05-07 09:53:32周春欢刘娟娟夏万松夏英万颖黄海
山东医药 2019年10期
周春欢,刘娟娟,夏万松,夏英,万颖,黄海
(1 贵州医科大学医学检验学院,贵阳550004;2 湖南医药学院检验医学院;3 贵阳中医学院第一附属医院;4广西壮族自治区南溪山医院)
胃癌的发生、发展是多因素、多基因参与的复杂过程[1],其病因和发病机制尚不完全明确。根治性手术是目前惟一有可能治愈胃癌的手段。但由于胃癌早期症状不典型,多数患者就诊时已属中晚期,错过了最佳手术时机,即使可以手术治疗,术后也极易复发和转移。因此,寻找新的肿瘤治疗方法迫在眉睫。长链非编码RNA(lncRNA)是一类转录本长度超过200 nt的RNA分子,它们并不编码蛋白,而是以RNA的形式在表观遗传、转录及转录后水平调控基因表达。起初lncRNA被认为是基因组转录的“噪音”,不具有生物学功能。目前已证实,lncRNA可参与X染色体沉默、基因组印记以及染色质修饰等调控过程。近年研究发现,lncRNA还可参与肿瘤细胞的增殖、迁移、凋亡等生物学过程[2,3]。核内小RNA宿主基因16(SNHG16)定位于人类染色体17q25.1,是最早在神经母细胞瘤中发现的一种lncRNA[4]。有研究报道,在结直肠癌、膀胱癌、乳腺癌组织中lncRNA SNHG16高表达,并可参与肿瘤的侵袭和转移[5~7]。本课题组前期研究发现,胃癌组织lncRNA SNHG16高表达,且其高表达与肿瘤进展和患者预后不良有关[8]。但lncRNA SNHG16能否调控胃癌细胞凋亡及其具体的作用机制尚不清楚。2017年11月~2018年7月,我们观察了敲低lncRNA SNHG16对胃癌细胞凋亡的影响,并探讨其可能的作用机制。现报告如下。
1 材料与方法
1.1 材料 人胃癌高分化细胞株AGS(以下称AGS细胞),购自中国科学院上海生命科学研究院细胞资源中心,由贵州医科大学医学检验学院中心实验室于液氮罐中保存。si-SNHG16序列:正义链5′-CCCAGUGUUGACUCACCAATT-3′,反义链5′-UUGGUGAGUCAACACUGGGTT-3′;阴性对照序列(si-NC):正义链5′-UUCUCCGAACGUGUCACGUTT-3′,反义链5′-ACGUGACACGUUCGGAGAATT-3′。si-SNHG16、si-NC序列均由上海吉玛制药技术有限公司设计合成。SNHG16、HPRT引物由本课题组前期设计[8],p53引物由Primer6.0软件设计。SNHG16、HPRT、p53引物均由上海生工生物工程股份有限公司合成。实时荧光定量PCR仪,瑞士Roche公司;流式细胞仪,美国Backman Coulter公司。LipofectamineTMRNAiMax转染试剂、Opti-MEM培养基,美国Invitrogen公司;Annexin V-FITC/PI凋亡检测试剂盒,江苏凯基生物技术股份有限公司;PrimeScriptTMRT Reagent Kit with gDNA Eraser逆转录试剂盒、SYBR Premix Ex TaqTMⅡ (Tli RNaseH Plus),日本TaKaRa公司;UNIQ-10柱式TRIzol总RNA抽提试剂盒,上海生工生物工程股份有限公司。p53、Bax兔源性单克隆抗体,美国Abcam公司;Cleaved Caspase-3兔源性多克隆抗体,美国CST公司;Bcl-2兔源性多克隆抗体,沈阳万类生物科技有限公司;GAPDH,南京巴傲得生物科技有限公司;辣根过氧化物酶标记的羊抗兔IgG二抗,杭州联科生物技术股份有限公司;Immobilon Western HRP底物化学发光液,美国Millipore公司;Gene Gnome凝胶成像分析系统,广州仪涛科学仪器有限公司;RIPA裂解液、Hoechst 33342染色液,北京索莱宝科技有限公司;Caspase-3活性检测试剂盒、BCA蛋白浓度测定试剂盒、Bradford蛋白浓度测定试剂盒,沈阳万类生物科技有限公司。
1.2 细胞分组处理 ①细胞培养:从液氮罐中取出冻存的AGS细胞,立即置于37 ℃水浴迅速融化,然后将细胞接种于含10% FBS、100 U/mL青霉素和100 μg/mL链霉素的RPMI 1640培养基中,置于37 ℃、5% CO2、饱和湿度的细胞培养箱中培养。待细胞融合85%左右时,按1∶3传代。②细胞转染:取传3代、对数生长期、生长状态良好的AGS细胞,用不含双抗的完全培养基轻轻吹打重悬,然后将细胞悬液接种于6孔板,每孔2 mL。随机将细胞分为空白对照组、阴性对照组、si-SNHG16组,每组设3个复孔。将6孔板置于37 ℃、5% CO2、饱和湿度的细胞培养箱中,当细胞融合50%~70%时,si-SNHG16组加入含si-SNHG16序列4 μL和LipofectamineTMRNAiMax转染试剂4 μL的Opti-MEM培养基,阴性对照组加入含si-NC序列4 μL和LipofectamineTMRNAiMax转染试剂4 μL的Opti-MEM培养基,空白对照组仅加入等量Opti-MEM培养基,转染6 h更换为新的无血清培养基。
1.3 各组lncRNA SNHG16、p53基因表达检测 采用qRT-PCR法。收集各组转染48 h细胞,使用UNIQ-10柱式TRIzol总RNA抽提试剂盒提取细胞总RNA,经琼脂糖凝胶电泳鉴定总RNA完整。按PrimeScriptTMRT Reagent Kit with gDNA Eraser逆转录试剂盒说明将总RNA逆转录为cDNA。以cDNA为模板,按SYBR Premix Ex TaqTMⅡ (Tli RNaseH Plus)说明进行PCR扩增。引物序列:SNHG16上游引物5′-GTGTAAGGATCTTCATGATG-3′,下游引物5′-GCTGGGAGCTAACTCACATT-3′,扩增片段长度163 bp;p53上游引物5′-CCACCATCCACTACAACTAC-3′,下游引物5′-CAAACACGCACCTCAAAG-3′,扩增片段长度136 bp;HPRT上游引物5′-CCTGGCGTCGTGATTAGTGAT-3′,下游引物5′-AGACGTTCAGTCCTGTCCATAA-3′,扩增片段长度131 bp。PCR反应体系共20 μL:SYBR Premix Ex TaqTMⅡ(Tli RNaseH Plus)10 μL,上下游引物各1 μL,灭菌双蒸水7 μL,cDNA模板1 μL;反应条件:95 ℃ 30 s,95 ℃ 5 s、60 ℃ 30 s、72 ℃ 30 s共40个循环。以HPRT为内参,采用2-ΔΔCt法计算目的基因相对表达量。实验重复3次,取平均值。
1.4 各组细胞凋亡检测 ①采用Annexin V-FITC/PI双染法流式细胞术。取传3代、对数生长期、生长状态良好的AGS细胞,按上述方法分组、转染。收集各组转染48 h细胞,用PBS润洗2次,吸弃PBS,用不含EDTA的胰酶消化1~2 min;加入适量完全培养基终止消化,移液枪轻轻吹打,用预冷的PBS调整细胞悬液密度为1×106个/mL,1 000 r/min离心5 min,尽可能吸除残余液体;加入Binding buffer 500 μL重悬,避光条件下依次加入Annexin V-FITC、PI各5 μL,充分混匀,室温反应30 min,1 h内上流式细胞仪检测细胞凋亡率。实验重复3次,取平均值。②采用Hoechst 33342染色法。取传3代、对数生长期、生长状态良好的AGS细胞,用巴氏吸管轻轻吹打成单细胞悬液,并接种于24孔板,每孔500 μL。次日,按上述方法分组、转染。各组转染48 h,吸除孔内培养基,用PBS润洗2次,每孔加入Hoechst 33342染液200 μL,然后将24孔板置于37 ℃、5% CO2、饱和湿度的细胞培养箱。孵育30 min左右,去除染液,PBS冲洗2次,吸弃PBS。将24孔板置于荧光显微镜下观察。每组随机选取5个200倍视野,计数每视野凋亡细胞数。实验重复3次,取平均值。
1.5 各组p53、Bax、Bcl-2、Cleaved Caspase-3蛋白表达检测 采用Western blotting法。取传3代、对数生长期、生长状态良好的AGS细胞,按上述方法分组、转染。收集各组转染48 h细胞,用预冷的PBS洗涤2次,3 500 r/min离心5 min,弃上清,加入适量的RIPA裂解液冰上裂解30 min,提取细胞总蛋白,经BCA法蛋白定量合格。然后加入5×SDS上样缓冲液,煮沸变性。取变性蛋白40 μg行SDS-PAGE(浓缩胶均为5%,p53、GAPDH分离胶为10%,Bax、Bcl-2、Cleaved Caspase-3分离胶为15%),恒压80 V跑胶约30 min,待蛋白进入分离胶时调整电压至120 V,当蛋白即将跑出分离胶时停止电泳。采用湿转法将蛋白电泳产物转印至PVDF膜上。5%脱脂牛奶封闭2 h,分别加入p53(1∶2 000)、Bax(1∶5 000)、Bcl-2(1∶500)、Cleaved Caspase-3(1∶1 000)、GAPDH(1∶20 000)一抗,4 ℃孵育过夜。次日,TBST洗膜10 min×3次,加入辣根过氧化物酶标记的羊抗兔IgG二抗(1∶20 000),室温孵育1 h。TBST洗膜10 min×3次,Immobilon Western HRP底物化学发光液发光、显影。采用Gene Gnome凝胶成像分析系统拍照,Image J软件分析各蛋白电泳条带灰度值。以GAPDH为内参,以目的蛋白电泳条带灰度值与内参蛋白电泳条带灰度值的比值作为目的蛋白相对表达量。实验重复3次,取平均值。
1.6 各组Caspase-3酶活性检测 取传3代、对数生长期、生长状态良好的AGS细胞,按上述方法分组、转染。收集各组转染48 h细胞,4 ℃下1 000 r/min离心5 min,弃上清。PBS洗涤1次,按上述条件再次离心,弃上清,加入适量RIPA裂解液冰上裂解15 min,提取细胞总蛋白,经Bradford法鉴定蛋白浓度合格。按照Caspase-3活性检测试剂盒说明配制反应体系,37 ℃孵育4 h,酶标仪于400 nm波长处检测各组吸光度(OD)值。以OD400值作为Caspase-3酶活性。实验重复3次,取平均值。
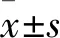
2 结果
2.1 各组lncRNA SNHG16、p53基因表达比较 si-SNHG16组、阴性对照组、空白对照组lncRNA SNHG16相对表达量分别为0.26±0.01、1.00±0.07、0.97±0.06,p53基因相对表达量分别为2.09±0.17、1.00±0.02、0.87±0.03。si-SNHG16组lncRNA SNHG16相对表达量明显低于阴性对照组和空白对照组(P均<0.05),p53基因相对表达量明显高于阴性对照组和空白对照组(P均<0.05),而阴性对照组与空白对照组lncRNA SNHG16、p53基因相对表达量比较P均>0.05。
2.2 各组细胞凋亡情况比较 Annexin V-FITC/PI双染法流式细胞术结果显示,si-SNHG16组细胞凋亡率为(7.19±1.16)%,阴性对照组为(2.99±1.02)%,空白对照组为(3.20±1.30)%。si-SNHG16组细胞凋亡率明显高于阴性对照组、空白对照组(P均<0.05),而阴性对照组与空白对照组比较P>0.05。
Hoechst 33342染色法结果显示,si-SNHG16组、阴性对照组、空白对照组细胞凋亡数分别为(18.8±3.1)、(6.8±2.3)、(4.6±1.1)个。si-SNHG16组细胞凋亡数明显高于阴性对照组、空白对照组(P均<0.05),而阴性对照组与空白对照组比较P>0.05。
2.3 各组p53、Bax、Bcl-2、Cleaved Caspase-3蛋白表达比较 见表1。

表1 各组p53、Bax、Bcl-2、Cleaved Caspase-3蛋白相对表达量比较
注:与空白对照组比较,*P<0.05;与阴性对照组比较,#P<0.05。
2.4 各组Caspase-3酶活性比较 si-SNHG16组、阴性对照组、空白对照组Caspase-3酶活性分别为1.40±0.06、1.00±0.02、0.93±0.02。si-SNHG16组Caspase-3酶活性明显高于阴性对照组、空白对照组(P均<0.05),而阴性对照组与空白对照组比较P>0.05。
3 讨论
据全球癌症报告显示,2018年预计全球新发胃癌约103.37万例,死亡约78.27万例,其发病率、病死率分别居所有恶性肿瘤的第5位和第3位[9]。我国是胃癌高发国家,每年新发病例41万左右,约占全球新发病例的40%[10]。由于胃癌起病隐匿,多无典型临床表现,大多数患者就诊时已处于中晚期,错过了根治性手术的最佳时机,因此临床治疗效果不甚满意。
lncRNA是一类不编码蛋白质,但具有广泛基因表达调控作用的非编码RNA分子,其可通过表观遗传、转录及转录后水平参与多种生命活动的调控过程[3]。近年研究发现,在多种肿瘤组织中lncRNA异常表达,在促进和维持肿瘤进展中发挥重要作用,已成为肿瘤领域的研究热点[11~13]。一般认为,lncRNA的作用具有4种模式,即信号传导、分子阻断、引导作用、中心平台,通过这4种模式的相互作用,参与基因的表观遗传调控和转录或转录后调控[14,15]。SNHG16是近年在神经母细胞瘤中发现的一种lncRNA,定位于人染色体17q25.1。Feng等[16]研究发现,SNHG16可通过调控miR-98/STAT3/Wnt/β-catenin信号轴,参与膀胱癌的侵袭和转移;Han等[17]研究发现,SNHG16在非小细胞肺癌组织中高表达,其表达变化与肿瘤大小、TNM分期、淋巴结转移及患者预后密切相关。有研究还发现,SNHG16作为miRNA海绵吸附分子,可通过“ceRNA”机制与目标基因竞争性结合miRNA分子并调控靶基因,从而参与肿瘤的侵袭和转移[18~20]。肿瘤的发生与细胞凋亡调控失常密切相关。当细胞凋亡机制遭到破坏,会导致肿瘤不可控地生长[21~24]。但目前鲜见lncRNA SNHG16调控胃癌细胞凋亡的报道。本研究采用RNA干扰技术敲低lncRNA SNHG16,发现AGS细胞凋亡率和凋亡数均明显增加,提示lncRNA SNHG16可能参与胃癌细胞的凋亡调控。
p53作为“基因组的守卫者”,当DNA受损时,可使细胞生命周期停滞或启动细胞凋亡程序,从而抑制肿瘤形成[25~28]。Bcl-2家族在细胞凋亡信号通路中具有重要作用。Bcl-2和Bax是Bcl-2家族最具代表性的凋亡调控因子。Bcl-2可通过阻止凋亡形成因子释放,如线粒体细胞色素C,从而抑制细胞凋亡;而Bax可通过与线粒体上的电压依赖性离子通道相互作用,促使凋亡形成因子释放,从而促进细胞凋亡[29,30]。有研究发现,p53可通过线粒体途径来诱导细胞凋亡,其机制可能与促进Bax表达、抑制Bcl-2表达有关[31,32]。本研究结果发现,si-SNHG16组p53表达明显高于空白对照组和阴性对照组,提示lncRNA SNHG16可能参与p53的调控。进一步研究发现,与空白对照组和阴性对照组比较,si-SNHG16组促凋亡蛋白Bax表达上调、抗凋亡蛋白Bcl-2表达下调。提示lncRNA SNHG16可能参与AGS细胞中Bax、Bcl-2凋亡相关蛋白的调控。Caspase-3是凋亡的关键执行者,可导致细胞结构的全面解体。在细胞凋亡早期阶段,Caspase-3被激活,转变成Cleaved Caspase-3,进而裂解相应的胞质或胞核底物,最终导致细胞凋亡。本研究结果显示,敲低lncRNA SNHG16,Caspase-3酶活性增加,Cleaved Caspase-3蛋白表达上调。提示lncRNA SNHG16还可通过调控细胞凋亡信号通路中Caspase-3酶活性,从而引起胃癌细胞凋亡。因此,我们认为lncRNA SNHG16可能直接或间接参与p53信号通路下游凋亡相关蛋白调控,从而介导胃癌细胞凋亡。
综上所述,敲低lncRNA SNHG16可促进胃癌细胞凋亡,其机制可能与激活p53信号通路下游凋亡相关蛋白表达,继而启动细胞凋亡程序有关。
猜你喜欢
看世界·学术下半月(2020年7期)2020-09-10 07:22:44
中国医学影像学杂志(2018年9期)2018-10-17 01:26:58
广东农业科学(2017年10期)2018-01-25 03:22:15
CHINESE JOURNAL OF AERONAUTICS(2017年1期)2017-11-21 12:54:14
中国医药指南(2017年3期)2017-11-13 02:55:17
法医学杂志(2015年4期)2016-01-06 12:36:36
法医学杂志(2015年4期)2016-01-06 12:36:36
医学研究杂志(2015年12期)2015-06-10 06:57:46
甘肃农业科技(2015年12期)2015-04-22 02:57:24
中国药理学通报(2014年2期)2014-05-09 08:22:30
