
第一性原理研究Be-S共掺杂AlN纳米片的电子结构和光学性质
2019-04-28 08:52屈艺谱刘玉怀
原子与分子物理学报 2019年5期
屈艺谱, 刘玉怀, 王 芳, 陈 雪
(1.郑州大学电子材料与系统国家国际联合研究中心,郑州 450001; 2.郑州大学 河南省电子材料与系统国际联合实验室,郑州 450001; 3.郑州大学 产业技术研究院,郑州450001; 4.郑州大学 信息工程学院电子与信息工程系,郑州450001)
1 引 言
近几十年来,石墨烯由于其优良的电子、光学、磁性等性能受到了人们的广泛研究[1-4]. 随着石墨烯材料在不同领域的成功应用,其他二维纳米材料的研究也逐渐展开,其中III族氮化物纳米材料引起了越来越多的关注[5、6]. 在二元III-V化合物中,AlN由于其出色的热力学、电子和光学性质而成为目前最吸引人的材料. 人们致力于开发基于AlN的器件应用的二维材料. 在纳米线[7]、纳米管[8]、纳米锥[9]、纳米带[10]和纳米片[11]等纳米材料的开发均有显著成效. 然而,缺陷问题是影响半导体材料物理和化学性质的主要原因之一,严重影响着半导体器件的应用. 大量的实验和理论证明,掺杂是一种改变半导体材料性质的有效手段. Ismail Yücel等人对AlN纳米片进行了Ga、P单掺杂和Ga-P共掺杂的研究,从光学角度分析了光学性质对于平行和垂直极化电场有各向同性[12]. 袁俊辉等人用第一性原理的方法研究了Ti,Cu和Zn掺杂AlN纳米片电磁性质,理论上证明了Ti掺杂AlN纳米片更适合用来制作稀磁半导体[13]. 掺杂对于AlN纳米片的电子结构,磁学特性和光学特性的影响研究还并不全面,通过对比不同元素掺杂有助于丰富已有的研究结论,同时有助于更好的理解掺杂的本质. 本文基于密度泛函理论广义梯度近似的平面波赝势(pseudo-potential plane-wave,PW-PP)方法研究了Be原子和S原子单掺杂和共掺杂AlN纳米片的电子性质和光学性质,对于掺杂体系进行了完善,同时对相关理论计算和实验具有一定参考意义.
2 理论模型和计算方法
依据石墨烯结构,构建二维AlN纳米片模型. 首先构造纤锌矿AlN5×5×5超胞. 空间群P63mc,设置初始晶格常数a=3.112 Å,c=4.982 Å,截取001面AlN单层优化得到5×5×1二维AlN纳米片,包含50个原子(25个Al原子,15个N原子). 为了避免相邻单元之间的层间相互作用,真空层设置为10 Å. 计算中选取的Al、N、Be,S价电子组态分别是Al-3s23p1、N-2s22p3、Be-2s2、S-3s23p4.
基于密度泛函理论的框架使用CASTEP(Cambridge Sequential Total Energy Package)软件包进行计算,交换互联函数采用广义梯度近似的Perdew-Burke-Ernzerhof[14]形式来处理(GGA-PBE). 在电子选项中,截断能设为450 eV,赝势为超软赝势,K点用Monkhorst-Pack[15]法设置网格为7×7×1. 在结构优化任务中,设置能量收敛精度1×10-5eV/atom,单原子最大力收敛精度0.03 eV/Å,最大应变收敛精度0.05 GPa,最大位移收敛精度0.001 Å.
3 结果和讨论
3.1 结构性质
图1(a)是优化后的本征AlN纳米片,Al-N键长1.892 Å,优化后为1.797 Å,这与其他人的数据十分接近[11]. 图1(a)(b)(c)(d)分别是本征、Be掺杂、S掺杂和Be-S共掺杂AlN纳米片的结构示意图,掺杂率分别为2%、2%、4%. 表1给出了优化后本征和掺杂AlN纳米片的键长和键角. 相比本征AlN纳米片,掺杂Be之后, Al-N键长增加,布局数减小,Al-N-Al(Be)键角几乎不变,整体结构变化不大,这是因为Be原子半径小于Al原子半径,同时Be-N共价特性强于Al-N的离子特性,成键能力更强. 掺杂S原子之后,Al-N键变化不大,Al-S键变长,Al-N(S)-Al键角增大,原因是键长的差异导致结构弯曲变形,S原子在Z轴正方向凸起,这种变形可以从图1(c)中看到. 对于Be-S共掺杂的情况,我们可以从表中看出,共掺杂中的Be-N键比Be单掺杂的键长更短,布局数更大,这意味着共掺杂中的Be原子和N原子的成键作用要更强. 同时Be-S键也比S单掺杂的Al-S键长短0.101 Å,布局数大0.06 eV,Al-S-Al(Be)键角增大,这样使得Be原子和S原子均向Z轴负方向移动.
3.2 电子结构
图2和图3所分别给出了掺杂的能带图和分波态密度图. 从图2(a)中看出,AlN是一种间接带隙材料,禁带宽度2.947 eV,与Sh. Valedbagi等人计算相符[11]. 图3(a)中显示,导带主要由Al3p和N2p,以及少量Al3s和N2s组成. 价带主要由N2p和Al3s3p,以及少量的N2s组成,价带顶位于对称点K处. 从图2(b)可以看出,掺杂Be原子后,仍为间接带隙材料,纳米片带隙宽度变化不大,费米能级进入价带顶,形成简并态. 单掺S的能带图如图2(c)所示,费米能级进入导带,形成杂质能级,出现简并态,表现出n型半导体特性,费米能级附近导带变化趋于平缓. 导带的宽度变窄,说明电子有效质量越大,非局域程度越小,能带的原子轨道延展性减弱. 从图3(c)可以看到,导带主要由Al3s3p、N2p和S3p组成,价带高能区主要由Al3s3p、N2p和S3p组成,低能区主要是Al3s3p、N2s和S2s组成. 同时0 eV处出现杂质态,主要由Al3s3p和S3p贡献. 从图2(d)中看出,带隙宽度为2.663 eV,相比图2(b)带宽减小,导带底和价带顶所处对称点位置不变. 图3(d)显示了Be-S共掺杂纳米片的总态密度和分波态密度. 结果表明,相比Be单掺杂,Be-S共掺杂导带的总态密度峰值降低,导带和价带整体向低能区移动的微弱趋势,同时受主能级所在价带区域略有展宽,价带中的空穴有效质量增加,非局域化程度明显. S原子的掺入使得Be原子的2s态和N原子的2p态轨道杂化程度增强,成键强度增加,这一点也可以从表1中Be-N键的布局数看出. 说明S原子的掺入提高了Be的掺杂浓度,减弱了原子间的排斥效用,可以起到激活Be原子的作用. 从以上分析可以看出,Be-S共掺杂有利于提高杂质原子的浓度和系统的稳定性,为AlN纳米片的有效掺杂提供了一种理论支持.

图1 AlN纳米片:(a)本征(b)掺杂Be(c)掺杂S(d)Be-S共掺杂Fig. 1 AlN nanosheet:(a) intrinsic (b) doped Be(c) doped S(d)Be-S codopding
表1 优化的本征和掺杂AlN纳米片的键长和键角
Table 1 Optimized Bond lengths and bond angles of intrinsic and doped AlN nanosheets

掺杂类型键的类型键长/Å键角类型键角/°布局数/eV本征AlNAl-N1.797Al-N-Al1201.82Be掺杂AlNAl-N1.813Al-N-Be122.0450.65Be-N1.702N-Be-N120.0050.62S掺杂AlNAl-N1.793Al-S-Al84.5420.64Al-S2.336N-Al-N120.6900.28Be-S共掺杂AlNAl-N 1.768N-Al-S119.7970.68Al-S2.222Al-S-Be104.2610.60Be-N1.607S-Be-N111.5650.76Be-S2.235N-Be-N136.3910.34
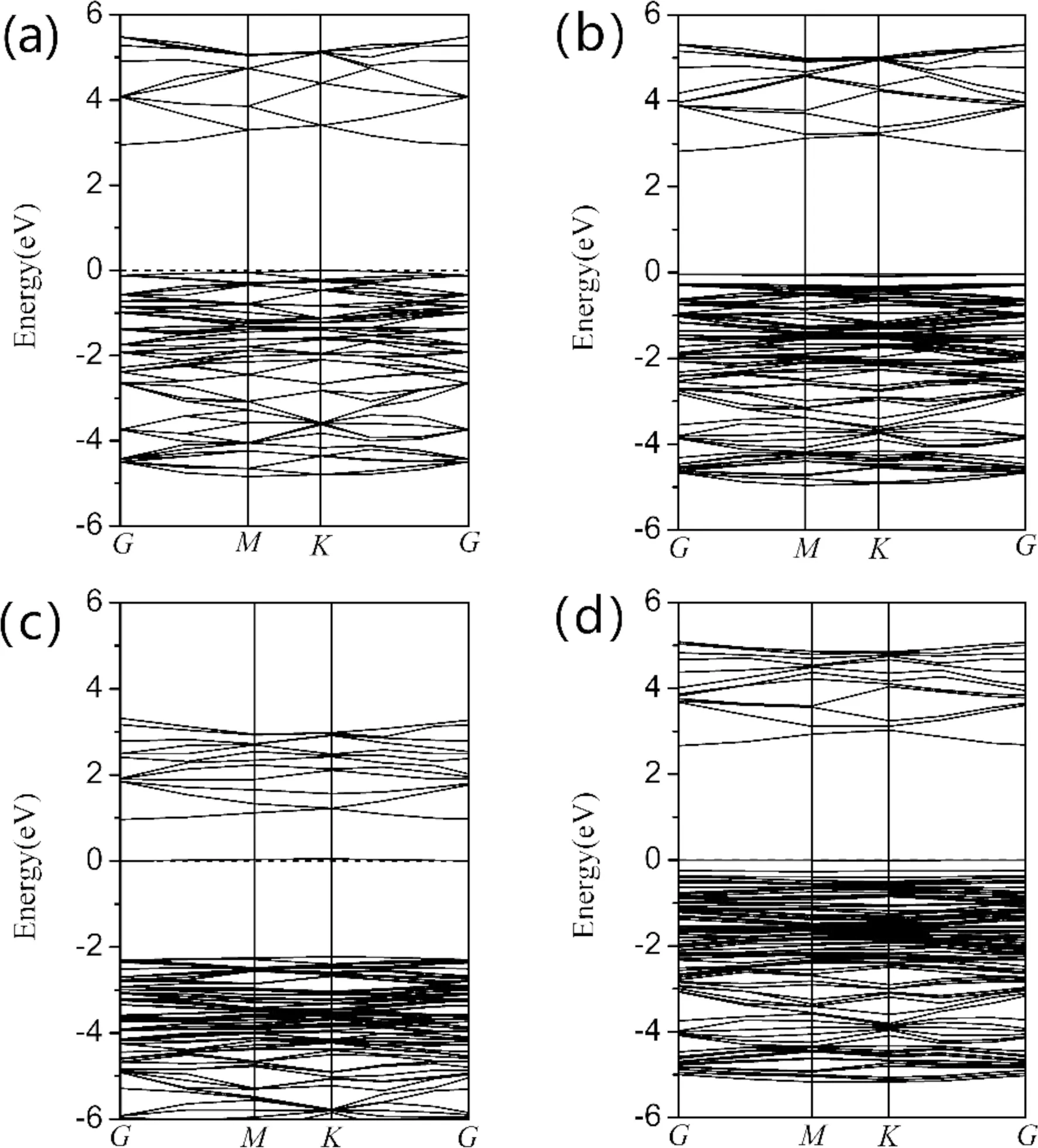
图2 能带:(a)本征(b)掺杂Be(c)掺杂S(d)Be-S共掺杂Fig. 2 Energy band:(a) intrinsic (b) doped Be(c) doped S(d) Be-S codoping
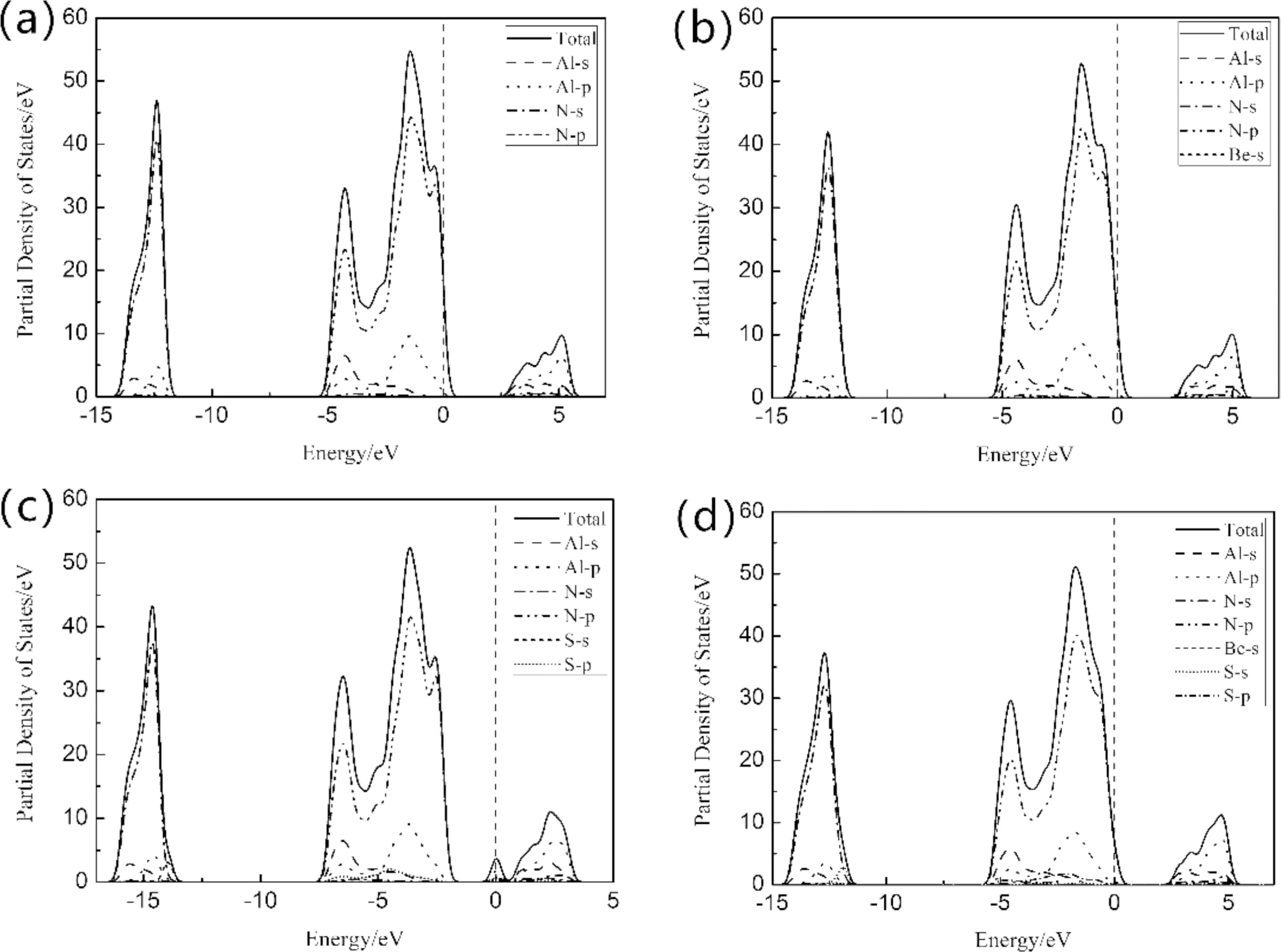
图3 分波态密度:(a)本征(b)掺杂Be(c)掺杂S(d)Be-S共掺杂Fig. 3 Partial density of states:(a) intrinsic (b) doped Be (c) doped S (d) Be-S codoping
3.3 差分电荷密度
为了分析原子之间的电荷存在转移情况,计算了掺杂AlN纳米片的差分电荷密度图,如下图4(a)(b)(c)(d)所示. 深色区域表示电子缺失,浅色区域表示电子富集. 从图4(a)看出,Al原子和N原子周围显示离子键成分,共同组成了共价键. Be替代Al掺杂后, Be原子和邻近N原子最高占据分子轨道发生轨道杂化,Be原子出现电荷转移,并在附近产生了浅的受主能级. 单掺杂S原子,Al原子和S原子之间成键作用减小,相比Al-N键,离子键特征不明显. 同时掺杂Be、S原子后,Be原子和S原子之间相互作用减小,离子键有断裂趋势,但是Be原子和邻近N原子的成键作用增强,说明S原子可以激活Be掺杂的AlN纳米片的作用,对周围原子产生了强的相互作用,对Be原子的掺杂浓度有所提升. 这一点从表1的布局数也可以看出.

图4 差分电荷密度:(a)本征(b)掺杂Be(c)掺杂S(d)Be-S共掺杂Fig.4 Differential Charge Density:(a) intrinsic (b) doped Be(c) doped S(d) Be-S codoping
3.4 光学性质
通过CASTEP光学运算模块对掺杂AlN纳米片的部分光学性质进行了计算,如复介电函数的实部和虚部,吸收谱和能量损失谱. 相关公式如下:
ε(ω)=ε1(ω)+iε2(ω)
(1)
ε1(ω)=n2-k2,ε2(ω)=2nk
(2)
(3)
(4)
3.4.1复介电函数
复介电函数作为沟通带间跃迁的微观物理过程和固体电子结构的媒介,反映了固体能带结构和其他信息. 图5(a)和(b)是不同掺杂类型的AlN纳米片复介电函数的实部和虚部. 图(a)中可看出,本征的AlN纳米片静态介电常数约为1.3 eV,在0~5 eV范围内,随着能量增加介电常数在5.1 eV处附近形成强的介电峰,在5.1~7.1 eV范围内迅速下降,7.1 eV之后变化缓慢并趋于稳定. 掺杂的AlN纳米片静态介电常数均高于本征结构,介电峰的能量范围向低能区移动,其中S掺杂的AlN纳米片在低能区0.85 eV处出现介电峰1.9. 虚部图像中,三种不同的掺杂类型和本征的AlN纳米片主峰峰值出现在同一位置,均在6.15 eV左右,S掺杂在低能区1.56 eV处有第二主峰,这是由于掺杂导致的纳米片结构发生变化. 掺杂类型的介电峰的能量范围相比本征纳米片略有展宽,这与能带图变化一致.
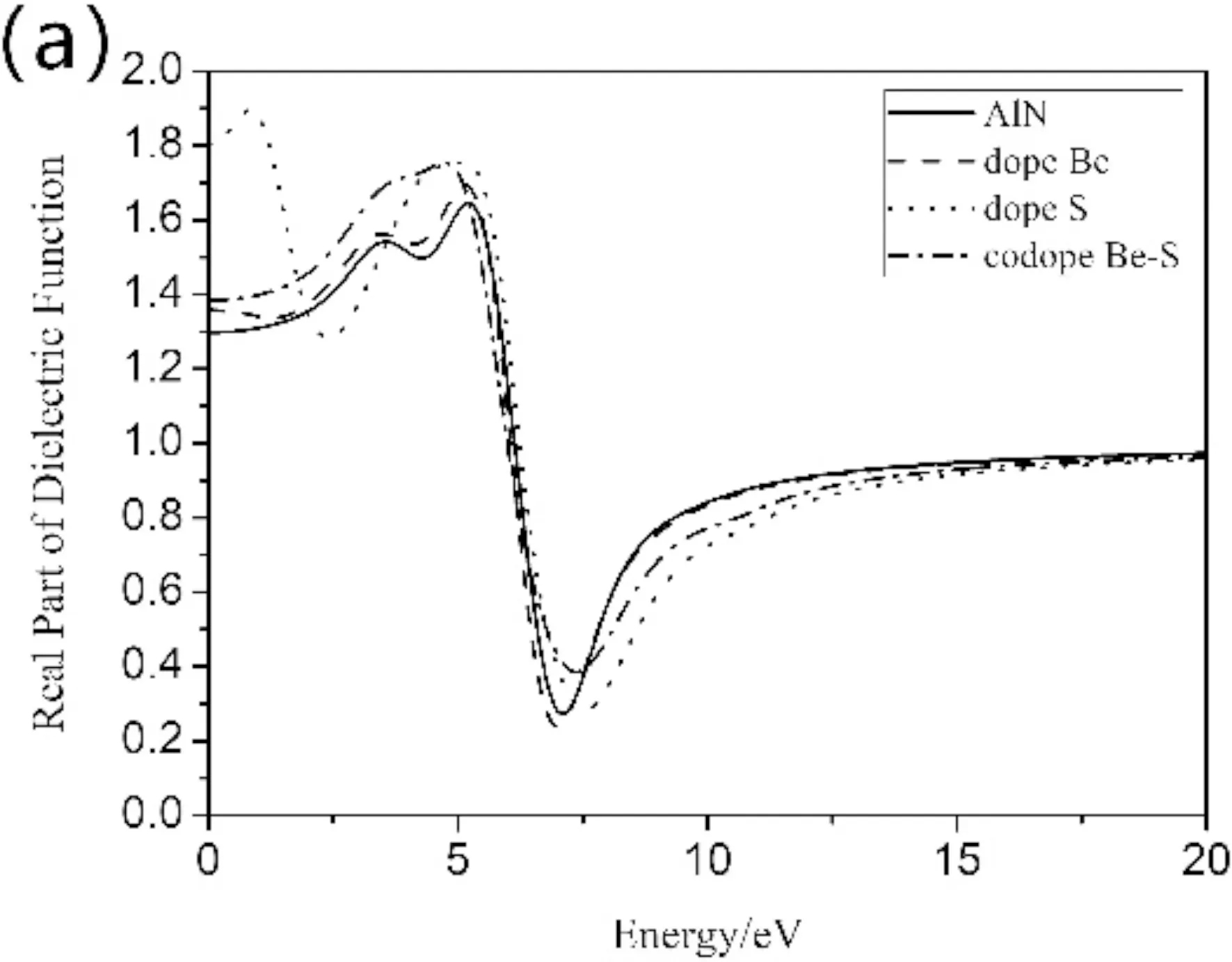

图5 复介电函数:(a)实部(b)虚部Fig.5 Complex dielectric function:(a) real part (b) imaginary part
3.4.2吸收谱和能量损失谱
本征和掺杂类型的AlN纳米片吸收谱和能量损失谱如图6(a)(b)所示. 本征AlN纳米片对能量的吸收范围在1.35~11.6 eV,在6.58 eV处有最大吸收系数5.9×104cm-1,低于1.35 eV和高于11.6 eV区域吸收系数几乎为0,表明体系是透光的. 掺杂导致的光吸收范围明显增宽,吸收峰峰值有有所增大,同时S掺杂使得低能区出现第二吸收峰,其他掺杂类型峰值出现的能量区基本不变. 电子能量损失函数(EELS)是描述材料中电子快速穿过的能量损失的重要因素,峰值与共振频率有关,称为等离子体频率. 图6(b)中,本征AlN纳米片在7.25 eV处有尖锐峰1.68,峰值所对应的等离子体边缘能量是指材料从金属到绝缘体的转变点. S掺杂在7.25 eV处也出现峰值,在低能区19 eV处出现第二高峰. Be掺杂和Be-S共掺杂导致峰值降低,能量范围变宽,并有向高能区移动微弱趋势.

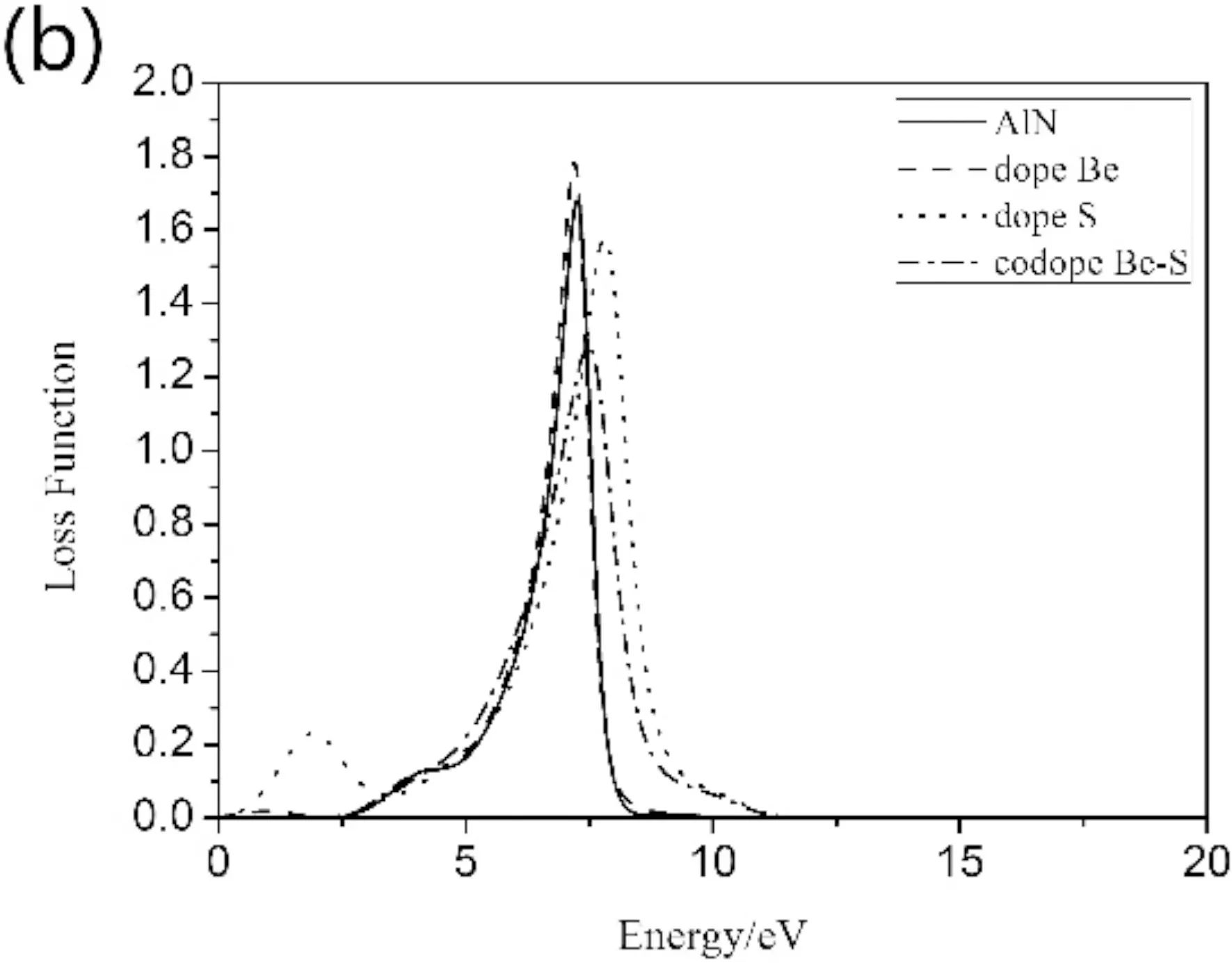
图6 (a)吸收谱(b)损失谱Fig.6 (a) absorption spectrum (b) loss spectrum
4 结 论
在本文中,我们使用基于第一性原理计算的CASTEP软件,分别对Be、S掺杂和Be-S共掺杂AlN纳米片的电子结构和部分光学性质进行了研究. 通过研究发现,Be掺杂对AlN纳米片的结构影响不大,而S掺杂和Be-S共掺杂改变了AlN纳米片结构,造成了S原子和Be原子在Z轴方向都有所弯曲移动. Be-S共掺杂比Be单掺杂更具有提高受主原子浓度,减小局域化特征等优点. 掺杂的AlN纳米片依然是间接带隙,且能带变窄. 同时,S原子掺杂的AlN纳米片显示有半金属特性,在复介电函数和吸收谱低能区出现介电峰,在特定范围内对红光等可见光有一定的吸收作用,这可能对制作红光探测装置有一定的帮助. 共掺杂的AlN纳米片对光的响应范围扩大,对高频率的光吸收强度增大. 这项工作为掺杂AlN纳米片在电子和光子器件中的应用提供了理论基础.
猜你喜欢
高中数理化(2022年16期)2022-09-14
装备维修技术(2021年36期)2021-10-25
弹箭与制导学报(2021年3期)2021-07-30
原子与分子物理学报(2020年3期)2020-05-15
青岛大学学报(工程技术版)(2019年2期)2019-09-10
中学生数理化(高中版.高考理化)(2019年6期)2019-06-22
网印工业(2019年4期)2019-05-21
重型机械(2019年2期)2019-04-28
西安工业大学学报(2019年2期)2019-04-02
吉首大学学报(自然科学版)(2018年3期)2018-07-03
