
氰基和氧改性g-C3N4吸附氧气的第一性原理研究
2019-04-28 08:53韩兴华王艳红焦纬洲
原子与分子物理学报 2019年5期
董 婷, 韩兴华, 陈 芳, 王艳红, 焦纬洲
(1.中北大学 超重力化工过程山西省重点实验室, 太原 030051; 2.中北大学 化学工程与技术学院, 太原030051)
1 引 言
类石墨相的碳氮化合物(g-C3N4)由于其稳定性高,容易制备,吸收可见光等优点成为光、催化领域的热点材料,比如光解水制氢[1]、光催化降解有机物及光催化有机反应等[2,3]. 然而g-C3N4层与层之间紧密堆积使得比表面积小,活性位点少且光生电子和空穴复合率高,这些严重限制了其光催化活性. 寻求改善g-C3N4催化性能的方法一直是科研人员对其研究的热点.
化学改性是一种改善g-C3N4电子结构和表面性质的有效方法. 通过向g-C3N4中掺杂金属、非金属元素,可提高g-C3N4的光催化效率[4]. Zhang等[5]制备了Fe/g-C3N4,金属原子镶嵌在g-C3N4结构单元中,改变了g-C3N4的电子结构. 在苯氧化为苯酚反应中,苯的转化率从5%提高到45%. 理论计算结果表明:Fe的引入使活性氧物种更容易生成,从而降低了反应的活化能,提高了反应的转化率. 阮等[6]的研究结果表明:B、P、S三种非金属掺杂的g-C3N4体系,相比g-C3N4,掺杂后体系的禁带宽度降低,拓展了其光吸收阈值,催化效率明显提高. Yu等[7]成功开发了一种新型的碱辅助合成方法,制备了富含氮空缺的g-C3N4纳米片,体相和表面的氮缺陷有利于拓展材料的可见光吸收范围以及提高光生载流子的分离能力,使得可见光下催化产氢速率大幅提升. Zhang等[8]制备了氰基改性的g-C3N4纳米层,用于可见光下催化苄胺偶联制亚胺,其催化剂活性达到g-C3N4的2.4倍,并且在苄胺衍生物的氧化偶联反应中展现出良好的活性和选择性. Wang等[9]制备了氧掺杂的g-C3N4催化剂并应用于析氢反应, 实验结果表明,O掺杂g-C3N4的光催化产氢速率(λ=800 nm)是g-C3N4(λ>420 nm)的25倍.
g-C3N4、氰基改性g-C3N4和氧掺杂g-C3N4在光催化分解水反应过程有很好的活性,也可用于氧还原反应[10]及氧化偶联反应[11]等. 表面氧化还是某些催化氧化反应的重要中间步骤,因此,有必要对氧气在催化剂表面吸附进行理论研究. 氧气在g-C3N4上吸附的研究有过报道,但氧气在氰基改性g-C3N4和氧掺杂g-C3N4上的吸附未见报道. 在此,本文中利用密度泛函理论计算对g-C3N4、氰基改性g-C3N4和氧掺杂g-C3N4的结构模型、禁带宽度和态密度进行系统研究,具体分析其光催化性能提升的原因. 此外,研究了氧气在其表面上的吸附行为.
2 计算方法
使用Material Studio7.0软件包中的Dmol3模块进行g-C3N4、氰基改性g-C3N4和氧掺杂g-C3N4几何结构和电子性质的计算. 为了消除层间距离内周期性的影响,使用了15 Å的真空距离将两个相邻的结构分隔开,a和b分别为16.7 Å和12.4 Å. 使用BLYP泛函的广义梯度近似(GGA)用于DFT交换相关能量,布里渊区采样3×3×4 k点.
吸附能计算公式见公式(1)[12]:Etotal为复合体系的总能,Eoxygen为单个氧气的能量,Eg-C3N4为吸附前平面g-C3N4的能量. 如果Eads<0,则这个吸附体系能够稳定存在,且吸附能越负,体系越稳定.
Eads=Etotal-Eoxygen-Eg-C3N4
(1)
3 结果与讨论
3.1 g-C3N4、氰基改性g-C3N4和氧掺杂g-C3N4的结构优化
根据文献报道的结构模型,在 Dmol3模块中分别设计出g-C3N4、氰基改性g-C3N4和氧掺杂g-C3N4结构模型,采用第2部分的方法对上述模型进行结构优化和禁带宽度的计算,优化后的结构模型和禁带宽度值见图1. 可看出,g-C3N4、氰基改性g-C3N4和氧掺杂g-C3N4的禁带宽度值分别为2.58、2.08和2.38 eV,改性后禁带宽度的绝对差值分别为0.5和0.2 eV. 其与相应文献的绝对差值(0.35和0.34 eV)有一定的误差[7,9],这可能是由计算软件和方法的不同造成. g-C3N4的禁带宽度为2.58 eV,与实验值2.7 eV较为接近,表明所建结构模型合理. 与g-C3N4相比,氰基改性和氧掺杂g-C3N4禁带宽度值减小,可提高其对可见光的吸收能力,并且大幅度降低光生空穴和电子的复合几率,进而提高催化剂的光催化活性.
3.2 g-C3N4、氰基改性g-C3N4和氧掺杂g-C3N4的态密度
图2为g-C3N4、氰基改性g-C3N4和氧掺杂g-C3N4的DOS和PDOS图,可看出,导带和价带的差值与计算所得禁带宽度值完全一致. 价带位置上移或导带位置下移均可使禁带宽度减小,从而使光催化活性提高. g-C3N4和氰基改性g-C3N4的价带主要是N2p轨道,导带是N2p和C2p轨道,这与文献报告的结果一致[7,13]. 氧掺杂g-C3N4的价带同样主要是N2p轨道,导带是N2p和C2p轨道,O2p轨道所占几率很小. 相对g-C3N4,氰基改性g-C3N4和氧掺杂g-C3N4的价带位置没有移动,导带位置均下移,但氰基改性g-C3N4的导带位置下移幅度较大,从而导致其禁带宽度值较小.
3.3 g-C3N4、氰基改性g-C3N4和氧掺杂g-C3N4上氧气的吸附能
g-C3N4、氰基改性g-C3N4和氧掺杂g-C3N4结构模型吸附氧气后,模型均由平面扭曲成波纹结构,氧气均以一定的角度吸附在催化剂上方. 根据公式(1)计算出每个体系的吸附能,结果见表1. 吸附后O-O键的键长同样见表1. 可看出,g-C3N4、氰基改性g-C3N4和氧掺杂g-C3N4的吸附能分别为0.37、0.35和0.37 eV;吸附后氧气的键长分别为1.245、1.245和1.245 nm. 表明,氧气在其表面的吸附为物理吸附并且不稳定.
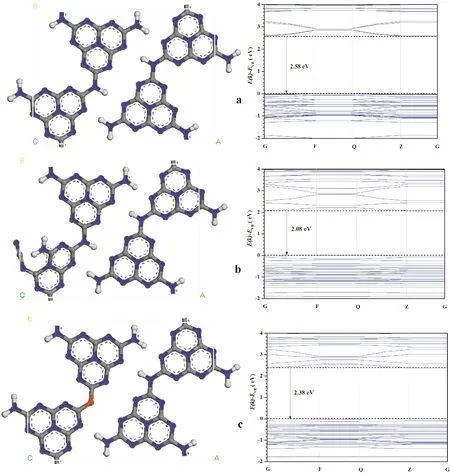
图1 g-C3N4(a)、氰基改性g-C3N4(b)和氧掺杂g-C3N4(c)的结构模型和禁带宽度(蓝色原子是N,灰色原子是C,白色原子是H,红色原子是O)Fig. 1 Structure models and calculated band structures of g-C3N4, g-C3N4 decorated with cyano and g-C3N4 doped with oxygen

图2 g-C3N4(a)、氰基改性g-C3N4(b)和氧掺杂g-C3N4(c)的DOS和PDOSFig. 2 DOS and PDOS of g-C3N4, g-C3N4 decorated with cyano and g-C3N4 doped with oxygen
表1 O2在g-C3N4、氰基改性和氧掺杂g-C3N4的吸附数据
Table 1 The adsorption data of oxygen on g-C3N4, g-C3N4decorated with cyano and g-C3N4doped with oxygen
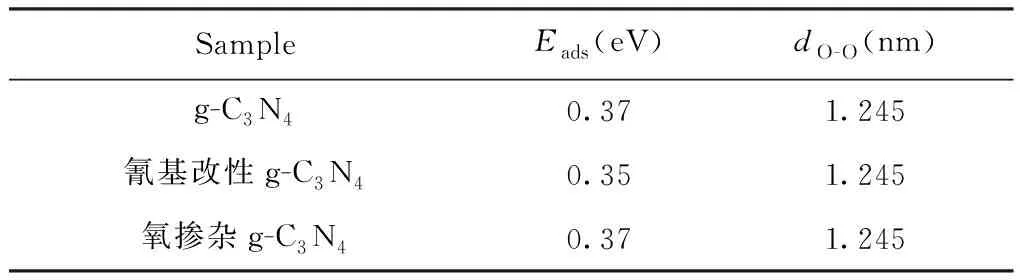
SampleEads(eV)dO-O(nm)g-C3N40.371.245氰基改性g-C3N40.351.245氧掺杂g-C3N40.371.245
4 结 论
在Dmol3模块中建立了g-C3N4、氰基改性g-C3N4和氧掺杂g-C3N4结构模型,计算所得禁带宽度值与实验值较为接近,可认为所建模型合理. 三种材料的价带主要是N2p轨道,导带是N2p和C2p轨道. 与g-C3N4相比,氰基改性g-C3N4和氧掺杂g-C3N4的价带位置没有移动,导带位置均下移,从而导致了禁带宽度变小. 氧气均以一定的角度吸附在三种材料的上方,且在表面的吸附为物理吸附.
猜你喜欢
装备维修技术(2021年36期)2021-10-25
弹箭与制导学报(2021年3期)2021-07-30
河南化工(2021年4期)2021-05-12
昆明医科大学学报(2021年2期)2021-03-29
原子与分子物理学报(2020年3期)2020-05-15
四川警察学院学报(2019年6期)2019-12-28
网印工业(2019年4期)2019-05-21
重型机械(2019年2期)2019-04-28
西安工业大学学报(2019年2期)2019-04-02
吉首大学学报(自然科学版)(2018年3期)2018-07-03
