
氧掺杂对钠在石墨烯上吸附性能的影响
2019-04-28 08:52张占东姚利花
原子与分子物理学报 2019年5期
张占东, 姚利花
(山西大同大学机电工程学院,大同 037003)
1 引 言
当今社会汽车需求与环境保护的矛盾日益突出,汽车的电动化趋势日益明显,二次电池凭借其优异的电化学性能,大量应用于电动汽车领域. 钠具有无污染、存量大、成本低的优点,而且它具有较高的电极电压(相对于标准氢电极,Eφ(Na+/Na) = -2.71 eV). 因此,钠离子电池更适合储存能量,有望成为新型二次电池. 现有的传统电极材料[1]已经越来越不能满足市场对二次电池日益增长的高比能量和高比功率需求. 目前,人们对新型二次电池负极材料的研究非常重视,其中,炭黑、[2]热解碳、[3]碳纤维[4]和石墨[5]等负极材料受到关注.
石墨烯由于其奇异的电子特性和独特的晶格结构受到高度重视. 石墨烯的电子迁移率非常高,且不会受温度变化的影响;石墨烯具有六角蜂窝状的二维平面结构,层间距较大,为钠原子提供了额外的储存位点,使钠原子在石墨烯层间很好的储存,克服了储钠容量受限问题. Yoo 等发现锂在石墨烯的储存量达到784 mAh/g,是锂在石墨中储存量的两倍,说明石墨烯具有高的储存能力[6].因此,石墨烯有望成为钠离子电池的负极材料.
第一性原理作为研究和预测材料性能的重要方法,近年来得到广泛的应用. 姚等[7-9]利用第一性原理研究了钠在石墨烯上的吸附性能;余等[10]利用第一性原理研究了压力下CaN2的结构稳定性和电子结构;欧等[11]利用第一性原理研究了单层MoS2表面吸附Ag6团簇的电子结构.
本文采用第一性原理研究钠原子在O-graphene上的吸附性能. 主要研究钠原子吸附在O-graphene的吸附能、电荷密度、态密度和储存量,并与P-graphene进行对比.
2 模拟方法和计算模型
2.1 模拟方法
采用基于密度泛函理论(DFT)的第一性原理计算方法,使用Materials Studio软件中的DMol3程序包. 电子交换关联势采用广义梯度近似(GGA)下的PW91泛函进行处理,自洽迭代过程简约布里渊区积分k点使用Monkhorst-Pack方法[12]选取2×2×1格点,在做自洽计算时总能量误差不大于1.0×10-4Hartree/atom. 计算中自旋不受限制,并且选择标准自旋为初始自旋,在倒易的k空间中,轨道截止选取5.1 Å,真空层取20 Å.
Na原子在石墨烯上的吸附能Ead:
Ead=[EGraphene+Na-(nENa+EGraphene)]/n
(1)
式中,EGraphene+Na是吸附Na原子后石墨烯的总能量,ENa是Na原子的总能量,EGraphene是吸附Na原子前石墨烯的总能量,n是Na原子的个数.
2.2 结构
如图1 (a)和(b)上图所示,是P-graphene和O-graphene的结构,考虑三个高对称吸附位置:H位于六方蜂巢格子正中心的上方;T位于碳或氧原子的正上方;B位于C—C键或C—O键中点的上方.
3 结果与讨论
3.1 吸附位置
如图1 (a)和(b)所示,在P-graphene和O-graphene中,把钠原子放在与石墨烯平面的垂直距离相等的高对称吸附位上进行结构优化. 表1列出了优化至最稳定结构时钠原子在各个吸附位置的吸附能Ead和钠原子与石墨烯平面的垂直距离d. 可以看出,两种石墨烯中,钠原子在H位上吸附时的吸附能最小,分别为-1.901 eV和-4.347 eV;且钠原子到石墨烯的垂直距离最小,分别为2.26 Å和2.16 Å. 因此,H位是最稳定的吸附位置,对于一个离子键的金属原子来说,它的平衡高度取决于金属原子和石墨烯相反电荷的吸引以及短程电子的排斥作用,[13]由于H位的电荷密度低于B位和T位(如图1 (a)和(b) Top view),金属原子更容易趋于稳定.
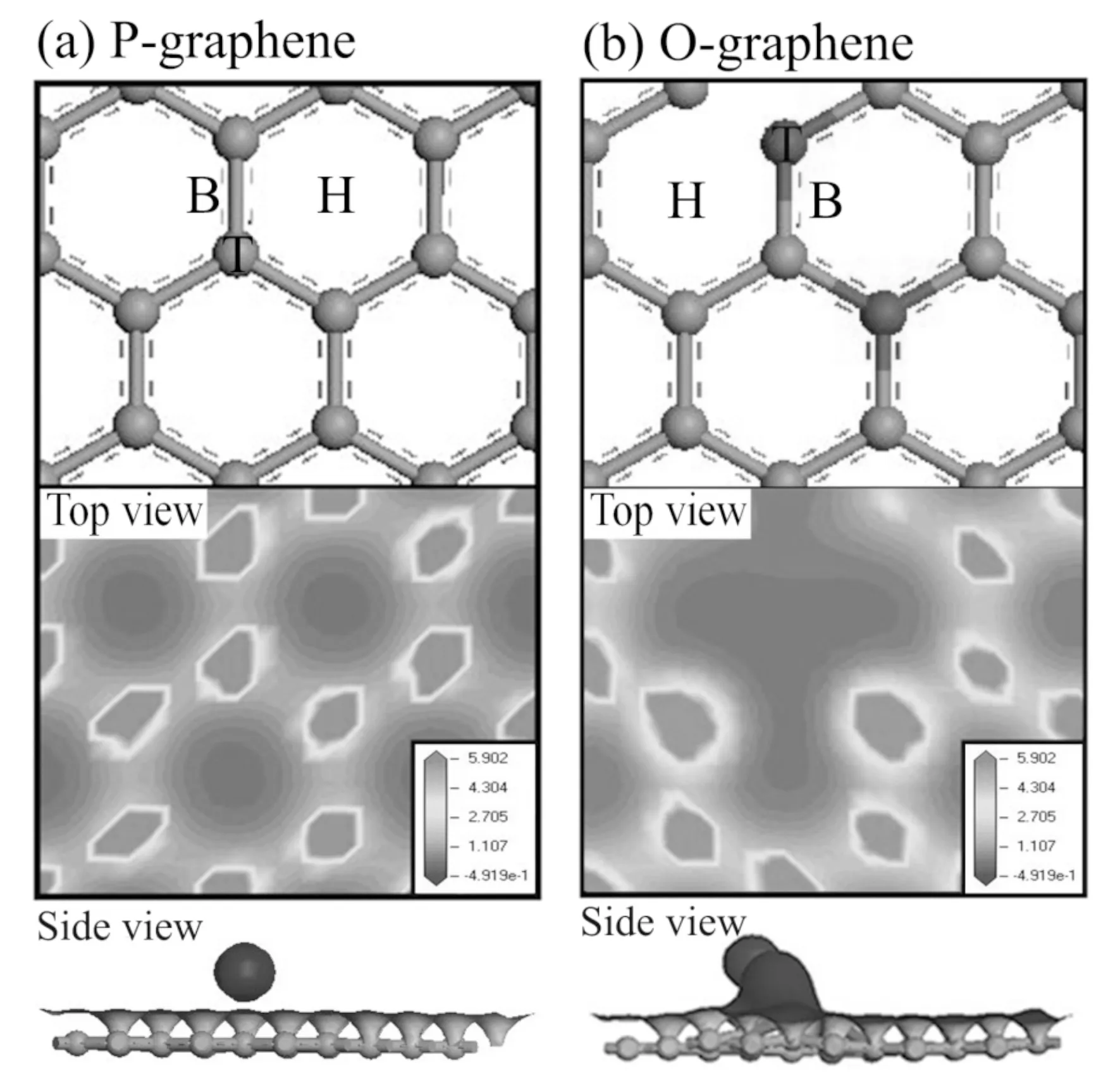
图 1 本征石墨烯和氧掺杂的石墨烯的结构及电荷密度. 灰色球代表碳原子,红色球代表氧原子. H、T、B代表吸附位置. Fig.1 The structures and charge densities of P-graphene and O-graphene. Grey spheres represent carbon atoms, red spheres represent oxygen atoms. H, T and B represent the adsorption sites.
表1 石墨烯上钠原子处于不同吸附位置时石墨烯对钠原子的吸附能(Ead)和钠原子与石墨烯平面的垂直距离(d).
Table 1 The adsorption energies (Ead) and the Na atom-graphene sheet distances (d) of different adsorption sites for the P-graphene and O-graphene.
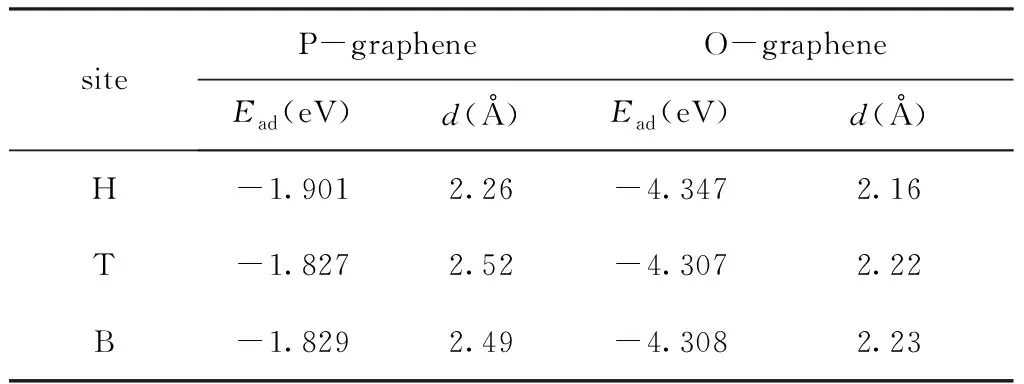
siteP-grapheneO-grapheneEad(eV)d(Å)Ead(eV)d(Å)H-1.9012.26-4.3472.16T-1.8272.52-4.3072.22B-1.8292.49-4.3082.23
3.2 电子结构
为了研究石墨烯对钠原子的吸附机制,计算了钠-石墨烯体系的电荷密度和态密度. 图1 (a)和(b)下图分别为P-graphene和O-graphene吸附钠原子后的电荷密度,如Side view所示,O-graphene与钠原子的电荷密度重叠严重,这会导致电子轨道的混合,使得电荷从钠原子传输到石墨烯更容易,因此,O-graphene与钠原子相互作用更强,这与Mpourmpakis 等[14]的报道相符合.
图2 (a)和(b)分别是P-graphene和O-graphene吸附钠原子后的分波态密度(PDOS). 可以看出:钠原子只对导带有贡献,这说明钠是完全离子化的,且其与石墨烯的相互作用力主要是库仑力[15];钠原子与P-graphene没有发生轨道杂化;O-graphene中,在费米能级以上1.462 eV和4.470 eV处,钠原子的3p轨道分别与氧原子的2p和碳原子的2p轨道发生杂化. 这是共价键的特征,[16]很好的解释了O-graphene和钠原子之间存在强的吸附作用,这与上述讨论结果相一致.

图 2 本征石墨烯和氧掺杂的石墨烯吸附钠原子后的分波态密度. Fig.2 The partial density of states (PDOS) of P-graphene and O-graphene after Na adsorption, the dashed line at zero indicates the Fermi level.
3.3 吸附量
钠离子电池电极材料的储钠量是非常重要的因素,为探究钠原子在石墨烯上的吸附性能,计算了钠原子在其上的吸附量. 图3 (a)和(b)分别是将10个钠原子放在P-graphene和O-graphene上优化后的结构. 可以看出,结构优化后,与两种石墨烯距离为3.883 Å的钠原子分别有7和10个,且分布散乱. 两种石墨烯吸附7和10个钠原子后,钠原子之间的平均距离分别为3.237 Å和3.335 Å,远远大于Na-Na键长1.91 Å;[17]钠原子的平均吸附能分别是-1.197 eV和-1.649 eV,小于钠原子的结合能-1.11 eV,[17]说明钠原子不会趋于团聚,不会导致电池充放电性能的降低. 因此,P-graphene对钠原子的吸附量是7,O-graphene对钠原子的吸附量是10,明显提高,并且都不会趋于团聚. 因此,O-graphene更适合储钠.
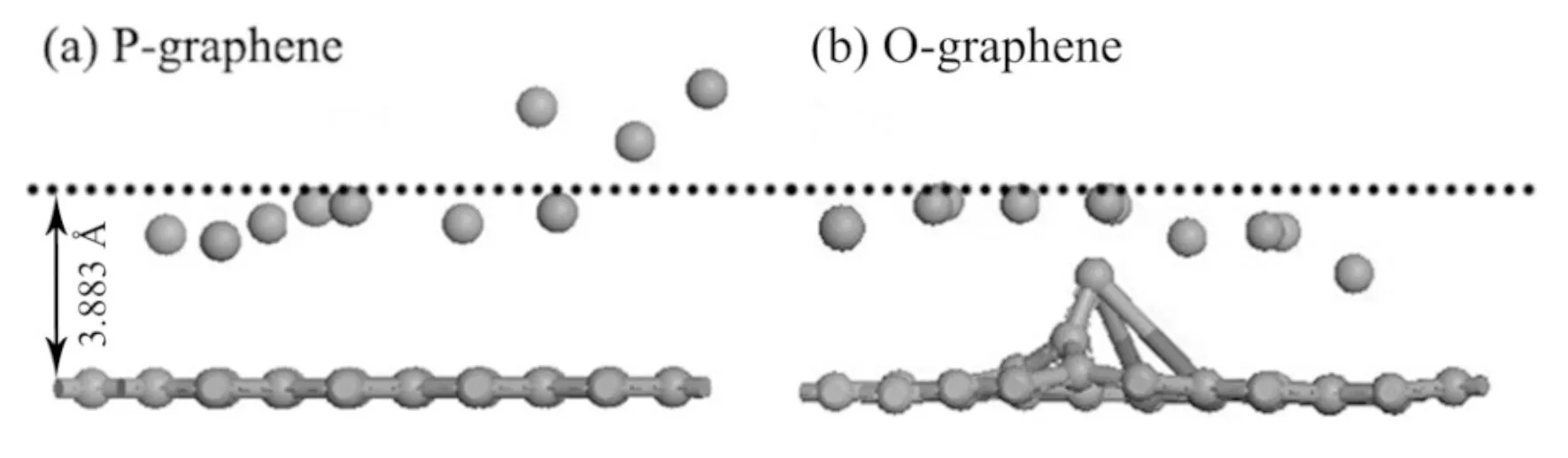
图 3 钠原子在本征石墨烯和氧掺杂的石墨烯的吸附结构. 灰色球代表碳原子,红色球代表氧原子,紫色球代表钠原子. Fig.3 The adsorption configurations of Na atoms adsorbed on P-graphene and O-graphene. Grey spheres represent carbon atoms, red spheres represent oxygen atoms, purple spheres represent sodium atoms.
4 结 论
本文研究了P-graphene和O-graphene对钠原子的吸附行为. 与P-graphene相比,O-graphene对钠原子的吸附能大大降低. O-graphene与钠原子发生轨道杂化,而P-graphene中不存在轨道杂化现象,说明钠原子与O-graphene的相互作用较大. 与P-graphene相比,O-graphene对钠原子的吸附量显著提高. 因此,O-graphene更适合储钠.
猜你喜欢
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
中学生数理化·中考版(2021年10期)2021-11-22
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
原子与分子物理学报(2020年5期)2020-03-17
燕山大学学报(2015年4期)2015-12-25
原子与分子物理学报(2015年3期)2015-11-24
原子与分子物理学报(2015年3期)2015-11-24
新高考·高一物理(2015年6期)2015-09-28
