
原发性纤毛运动障碍一例
2019-04-27 01:16杨俊俊徐兴祥王正东
中华肺部疾病杂志(电子版) 2019年4期
杨俊俊 徐兴祥 王正东 邹 晶
原发性纤毛运动障碍或称原发性纤毛不动综合征(primary ciliary dyskinesia, PCD)一种罕见的纤毛运动障碍的遗传性疾病,是第一个与纤毛运动障碍有关的人类疾病[1]。最近发现了超过40个与PCD有关的基因,每个基因都有许多致病突变,而不同的突变导致不同的临床和病理模式,从而对PCD的诊断造成挑战[2-3]。本院呼吸科于2014年6月收治一例PCD患者,现报道如下。
病例资料
患者,女,21岁。因“反复咳、痰、喘十余年,加重半年” 2014年6月11日入院。患者十余年来无明显诱因反复出现咳嗽,咳痰,为白黏痰,偶有黄绿痰,曾有痰中带血,色鲜红,伴胸闷,活动后气喘,无发作性喘鸣,偶有发热。十年前在外院诊断为“支气管哮喘”,予平喘、对症处理,但患者咳嗽、气喘可短暂缓解。近半年来患者咳嗽时轻时重,咯少量黄痰,胸闷、气喘加重,予抗感染、平喘等治疗可稍缓解。为进一步诊治收住入院。病程中饮食佳,大小便正常,精神、睡眠可。既往有“鼻窦炎”病史,于2011年“鼻窦炎”手术史。 无烟酒不良嗜好。月经史正常,未婚未育,父母非近亲婚配。家族无类似疾病史。
入院查体:T 36.3 ℃,P 80 bpm,R 20 bpm,BP 110/78 mmHg,神清,唇无紫绀,胸廓无畸形,双肺叩诊清音,双肺呼吸音粗,未闻及干湿性啰音,心率80次/min,律齐,各瓣膜听诊区未及病理性杂音,双手指、双足趾末端杵状改变,见图1。
辅助检查:(1)入院时检查:肺功能示:通气功能、CO弥散量基本正常。残气量、残总百分比中度增高。支气管舒张试验:可疑阳性,EFV1改善率11.3%,绝对值280 ml。PANCA、CANCA 、自身抗体系列(-)。肝肾功能正常。血常规:WBC 14.35×109/L,N 77.2%,PLT 329×109/L。抗酸杆菌(-)。痰细菌培养:铜绿假单胞菌。乙肝、戊肝、梅毒、HIV抗体阴性。鼻窦CT(2011年)显示鼻腔黏膜增厚,单组或多组鼻窦炎性改变,窦壁骨质增生硬化,伴鼻息肉改变,见图2。胸部HRCT提示支气管扩张,考虑“支气管扩张伴感染”,见图3;(2)支气管镜检查:气管内大量黄白色分泌物,隆突锐。左右肺:双侧气道内大量黄白色分泌物。吸取气管支气管分泌物及刷检后送取病原学检测:真菌、肺孢子菌、放线菌、奴卡氏菌、分枝杆菌均(-),抗酸染色(-);细菌培养:铜绿假单胞菌,肺泡灌洗液行结核分枝杆菌复合群DNA(-),非结核分枝杆菌复合群DNA(-)。支气管黏膜活检标本电镜:支气管黏膜上皮细胞纤毛普遍短小,横断面可见纤毛微管排列基本正常,部分外动力臂缺失,内动力臂完全缺失,超微结构符合纤毛不动综合征表现,合并纤毛发育不良可能。
最后诊断为原发性纤毛运动障碍。治疗及转归为予抗纤维化、抗感染、扩张支气管改善气道高反应性等治疗,患者痰量明显减少,偶感胸闷。定期随访。

图1 患者杵状指、杵状趾改变
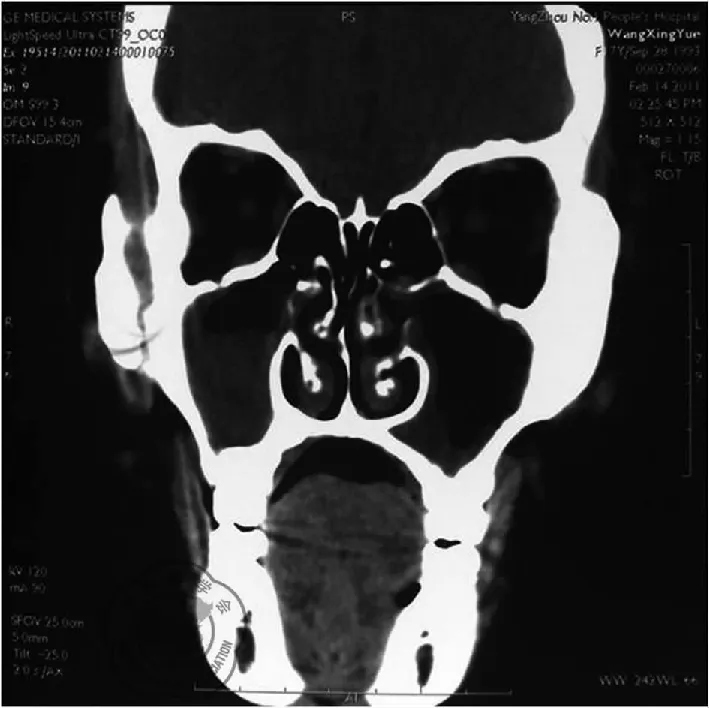
图2 鼻窦CT(2011年)显示鼻腔黏膜增厚,单组或多组鼻窦炎性改变,窦壁骨质增生硬化,伴鼻息肉改变
讨 论
原发性纤毛运动障碍(primary ciliary dyskinesia, PCD)是因纤毛结构缺陷导致活动障碍,引起纤毛清除功能降低及其他功能缺陷的常染色体隐性遗传病。同时合并支气管扩张、内脏转位、鼻窦炎/鼻息肉者,称为Kartagener综合征(Kartagener syndrome)。Shapiro等[4]的一项前瞻性研究发现,767位参与者,其中典型PCD患者305例,分别有143(46.9%),125(41.0%)和37 (12.1%) 的患者存在内脏正位(situs solitus, SS)、完全性内脏逆位(situs inversus, SI)和内脏对称位(SA)。而内脏异位人群PCD的患病率未知,有研究调查发现,在49位内脏异位受访者中,37%的受访者特征提示PCD[5]。一系列的偏侧缺陷被确定为典型PCD,包括2.6%和2.3%的SA合并复杂或简单的心脏缺陷,4.6%的SA者没有心脏缺陷,而合并SA的典型PCD者亦有较高的PCD相关的呼吸系统症状,如全年湿性咳嗽、全年的鼻充血、新生儿呼吸窘迫及杵状指和较低鼻一氧化氮(nasal nitric oxide, nNO)水平[4]。PCD的人群发病率约1/30 000~1/60 000。多于婴幼儿期发病,多年后确诊,父母多有近亲婚配史。研究认为,基因突变导致纤毛功能的缺陷,引起纤毛运动不良[6]。有研究者在英国老式牧羊犬中发现一种可能与PCD相关的新的致病基因突变,更多可能与PCD相关的基因突变或缺陷正被发现[7-10]。目前已有超过40个与PCD有关的基因被发现[2,11]。有研究认为PCD可能与人体免疫缺陷有关(humoral immunodeficiency, HID),但其内在联系尚有待进一步研究证实[12]。纤毛分布于多个组织器官,如呼吸道、胃肠道、耳道、输卵管、输精管、精子鞭毛、脑室等,纤毛的结构缺陷常引起多个系统受累的症状,常以呼吸道最严重[13]。有研究者发现与健康对照者相比,即使稳定的PCD亦有很高的呼吸暂停障碍,而睡眠呼吸障碍可加重PCD的肺部损害,从而提醒PCD患者家属注意监测患者肺功能[14]。2016 ERS及2018ATS指南中均强调目前PCD临床表现往往不典型,易误诊,其诊断缺乏金标准,诊断主要依据临床病史、nNO测定、基因检测及纤毛功能和超微结构的分析[15-16]。指南均提出了指导临床医生进行PCD诊断的程序,在进行PCD诊断测试前要求临床医生在评估患者时至少识别出4个PCD关键临床特征中的2个强的临床表型(其中包括作为足月婴儿原因不明的新生儿呼吸窘迫、全年每日咳嗽或鼻塞开始于六月龄前、器官侧性缺陷)。而后再逐步进行nNO检测、高速视频纤维分析(high-speed video microscopy analysis, HSVA)、透射电镜(transmission electron microscopy, TEM)下纤毛超微结构分析、基因检测。电镜下纤毛或鞭毛结构异常主要表现为:内、外动力臂缺失或异常;微管缺失或存在额外微管;链接体或轮辐的缺陷;纤毛的方向紊乱[17]。本例患者青年女性,父母非近亲婚配,病史多年,临床主要表现为反复发生的咳嗽、咳痰、呼吸困难、呼吸道感染、支气管扩张等,并发鼻窦炎,不伴内脏转位(右位心等)。体征表现为双肺呼吸音粗,曾有反复的双下肺干、湿性啰音,四肢表现杵状指和杵状趾。血常规联合影像学提示肺部炎症,伴有支气管扩张的卷发征、硬戒征、轨道征表现;鼻窦CT提示鼻窦炎;肺功能换气功能异常;痰培养、痰涂片明确感染病原菌;支气管黏膜电镜符合纤毛不动综合征表现。诊断“PCD”。值得注意的是PCD在临床有时与弥漫性泛细支气管炎(diffuse panbronchiolitis, DPB) 的鉴别较困难。本例患者病程中,曾一度误诊为“支气管扩张伴感染”,曾高度考虑DPB,予大环内酯类治疗无效。

图3 胸部HRCT示:两肺纹理增多,两肺下叶见网格影及点状、磨玻璃样稍高密度影,右肺中叶、两下肺病变见多发系轨道征,部分支气管囊状扩张、腔内见小液平
DPB是一种弥漫存在于两肺呼吸性细支气管的气道慢性炎症性疾病。受累部位主要是呼吸性细支气管以远的终末气道[18]。由于炎症病变弥漫性地分布并累及呼吸性细支气管壁的全层,故称之为弥漫性泛细支气管炎,突出的临床表现是咳嗽、咳痰和活动后气促,严重者可导致呼吸功能障碍,临床上易与其他慢性气道疾病混淆。检索文献发现临床曾报道2例PCD合并DPB者,有研究者认为DPB可能是PCD的肺部表现,而其中的内在联系尚有待进一步研究证实[19-20]。
猜你喜欢
中国神经精神疾病杂志(2022年3期)2022-07-14
中国临床医学影像杂志(2022年2期)2022-05-25
医学研究生学报(2021年4期)2021-12-02
辐射研究与辐射工艺学报(2021年2期)2021-05-06
临床与实验病理学杂志(2021年3期)2021-04-25
实用皮肤病学杂志(2020年2期)2020-05-11
散文诗世界(2019年6期)2019-09-10
意林·全彩Color(2019年7期)2019-08-13
家庭科学·新健康(2018年11期)2018-11-16
中西医结合心血管病电子杂志(2016年9期)2016-11-16
