
GSK3β在HGF信号通路中对改善心肌缺血再灌注损伤的作用研究*
2019-04-26 02:52潘艳艳史斌浩马梦晴林先和
重庆医学 2019年13期
潘艳艳,史斌浩,马梦晴,林先和
(安徽医科大学第一附属医院心血管内科,合肥 230022)
心血管疾病(cardiovascular diseases,CVD)仍是世界范围内导致患者死亡的主要原因,包括冠状动脉疾病、冠心病和缺血性心脏疾病,已成为全球性公共健康问题。随着经济的发展和生活水平的提高,人们生活作息和饮食习惯的改变,心肌梗死(myocardial infarction,MI)的发病率呈上升趋势[1]。临床上以改善缺血,尽早恢复心肌血流灌注,减少MI面积为治疗的首要原则[2]。大量研究表明,在缺血期除了细胞凋亡、坏死等促细胞死亡的机制外,还存在着细胞自噬(autophagy)等促细胞存活机制[3]。自噬是一种进化上保守的细胞内自我保护机制,细胞内多余的蛋白质聚集体和受损细胞器被隔离到自噬小泡中,随后与溶酶体融合降解[4]。在缺血状态下,心肌细胞可以通过不同的信号通路调控自噬系统,自噬的发生可促进对受损蛋白、脂质和细胞器等废物的清除[3]。因此,自噬被认为是心肌细胞对抗缺血性损伤的主要保护机制。既往研究表明,肝细胞生长因子(HGF)与其受体(Met)的结合可以激活下游信号通路促进自噬,与多种组织疾病自噬密切相关,如肺癌、结肠直肠癌、高糖性肾足细胞损伤等[5]。糖原合成激酶3β(GSK3β)是HGF下游通路中的一个重要通路蛋白,其活性的抑制可以激活多种下游通路对抗心肌缺血再灌注损伤引起的炎性反应,钙超载和氧化应激反应等[6]。有研究发现抑制GSK3β可以激活不同的信号通路,如腺苷单磷酸激活蛋白激酶(AMPK)通路、Beclin-1通路,可以调控多种疾病自噬[7-8]。然而,GSK3β在HGF促进自噬对抗缺心肌血再灌注损伤中的作用少见报道。本研究通过建立立体大鼠心肌细胞缺血再灌注模型,观察内源性HGF和磷酸化GSK3β(pGSK3β)的表达情况及下游通路蛋白和自噬相关蛋白的表达水平,研究HGF通过促进GSK3β的磷酸化调控自噬在减轻大鼠心肌细胞缺血再灌注损伤中的相关作用。
1 材料与方法
1.1主要材料 AMPK、肝激酶B1(LKB1)、HGF抗体(美国Affinity Biosciences公司);LC3Ⅱ抗体(美国Abcam公司);HRP-羊抗小鼠IgG、HRP-羊抗兔IgG、 β-actin、蛋白激酶B(Akt)抗体(上海Bioss公司);腺病毒(上海Hanbio生物科技有限公司);超敏型ECL发光液(美国 Advansta公司);胎牛血清(上海双洳生物科技有限公司)。
1.2方法
1.2.1细胞系的选择及培养 选用大鼠离体心肌细胞H9c2细胞系,90%高糖培养基(DMEM)、10%小牛血清及1%双抗(青霉素、链霉素)配成的培养基置于37 ℃ 5%CO2环境下的细胞培养箱中进行培养,取生长良好的细胞种植于6孔细胞培养板,继续培养备用。
1.2.2实验模型制备 实验分为正常对照组和实验组,实验组分别为缺血再灌注(I/R)组、I/R+HGF组、I/R+HGF+Ad-GFP组(不表达GSK3β的重组腺病毒Ad-GFP)、I/R+HGF+GSK3β-Ad-wt组(野生型GSK3β腺病毒GSK3β-Ad-wt)、I/R+HGF+GSK3β-Ad-S9A组[表达GSK3β不可磷酸化的组成型活性突变体,其第9位丝氨酸残基突变(Ser9)为丙氨酸,从而不能再被磷酸化失活而具有固有活性的GSK3β腺病毒GSK3β-Ad-S9A]和I/R+HGF+GSK3β-Ad-K85A组(表达催化失活的GSK3β复制缺陷型腺病毒载体,其第85位赖氨酸残基突变为甲硫氨酸,从而持续失活的GSK3β腺病毒GSK3β-Ad-K85A)。腺病毒感染方法为细胞贴壁于6孔细胞培养板,吸去原有培养基,加入1 mL新鲜培养基,选定多重感染(MOI)为100单位对应的病毒原液体积加入细胞中,摇匀。放入37 ℃培养箱中感染2 h,吸去含病毒的培养液,PBS清洗2次,加入新鲜的培养液。HGF激活剂组加入HGF激活剂(20 ng/mL),放入培养箱中培养1 h后更换新鲜培养液2 mL,继续培养24 h后制备I/R模型。分别吸出6组实验组中的完全培养基,加入2 mL缺血模拟液(NaCl 98.5 mmol/L,KCl 10 mmol/L,NaH2PO40.9 mmol/L,NaHCO36.0 mmol/L,CaCl21.8 mmol/L,MgSO41.2 mmol/L,乳酸钠40 mmol/L,HEPES 20 mmol/L)并置于低氧细胞培养箱(95% N2,5% CO2,37 ℃)中培养12 h,吸出缺血模拟液,加入2 mL完全培养基于普通培养箱中继续培养1 h。
1.2.3检测指标 采用Western blot检测Akt、GSK3β、LKB1、AMPK、LC3Ⅱ、Beclin-1表达。 模型制备完后,将培养板置于冰盒上,用预冷的PBS清洗2次,吸尽PBS,加入裂解液摇床上裂解30 min,用细胞刮刮至1.5 mL的EP管中,4 ℃ 12 000 r/min离心30 min,轻轻吸取上清液加入其1/4的SDS上样缓冲液,沸水中煮10 min,进行十二烷基磺酸钠-聚丙烯酰胺(SDS-PAGE)凝胶电泳,之后将蛋白转于聚偏氟乙烯(PVDF)膜上。聚偏氟乙烯(PVDF)膜用含50 g/L脱脂奶粉的TBST溶液封闭3 h。将含有蛋白的聚偏氟乙烯(PVDF)膜置于1∶1 000或1∶300的目标蛋白抗体的一抗稀释液中4 ℃过夜孵育,TBST冲洗3次,每次15 min,分别使用对应的1∶10 000二抗孵育1 h,TBST冲洗3次,每次15 min。最后用超敏型ECL发光液进行曝光显影,显影仪Bio-Rad成像系统拍照,Image J软件分析蛋白的相对表达量。

2 结 果
2.1各组GSK3β、pGSK3β表达情况 与对照组和I/R组比较,I/R+HGF组GSK3β的总表达量差异有统计学意义(P<0.05),而pGSK3β的表达量明显增加(P<0.05)。在HGF激活剂的刺激下,各GSK3β感染组的GSK3β的总表达量差异有统计学意义(P<0.05),I/R+HGF+GSK3β-Ad-wt组的GSK3β总表达量较低(P<0.05)。I/R+HGF+GSK3β-Ad-wt组和I/R+HGF+GSK3β-Ad-K85A组的pGSK3β明显升高(P<0.05),而I/R+HGF+GSK3β-Ad-S9A组降低(P<0.05),见图1。

1:对照组;2:I/R组;3:I/R+HGF组;4:I/R+HGF+Ad-GFP组;5:I/R+HGF+GSK3β-Ad-wt组;6:I/R+HGF+GSK3β-Ad-S9A组;7:I/R+HGF+GSK3β-Ad-K85A组;a:P<0.05与对照组比较;b:P<0.05,与I/R组比较;c:P<0.05,与I/R+HGF组比较
图1 各组GSK3β、pGSK3β表达情况
2.2各组LKB1、AMPK表达情况 与对照组比较,I/R组的LKB1和AMPK的表达量明显升高(P<0.05)。与I/R组相比,I/R+HGF组LKB1和AMPK的表达量明显升高(P<0.05)。在HGF激活剂的作用下,I/R+HGF+GSK3β-Ad-wt组和I/R+HGF+GSK3β-Ad-K85A组的LKB1和AMPK明显升高(P<0.05),而I/R+HGF+GSK3β-Ad-S9A组降低(P<0.05),见图2。
2.3各组LC3Ⅱ、Beclin-1表达情况 与对照组比较,I/R组LC3Ⅱ、Beclin-1的表达量明显升高(P<0.05)。与I/R组相比,I/R+HGF组LC3Ⅱ、Beclin-1的表达量明显升高(P<0.05)。在HGF激活剂的作用下,I/R+HGF+GSK3β-Ad-wt组和I/R+HGF+GSK3β-Ad-K85A组的LC3Ⅱ、Beclin-1明显升高(P<0.05),而I/R+HGF+GSK3β-Ad-S9A组降低(P<0.05),见图3。
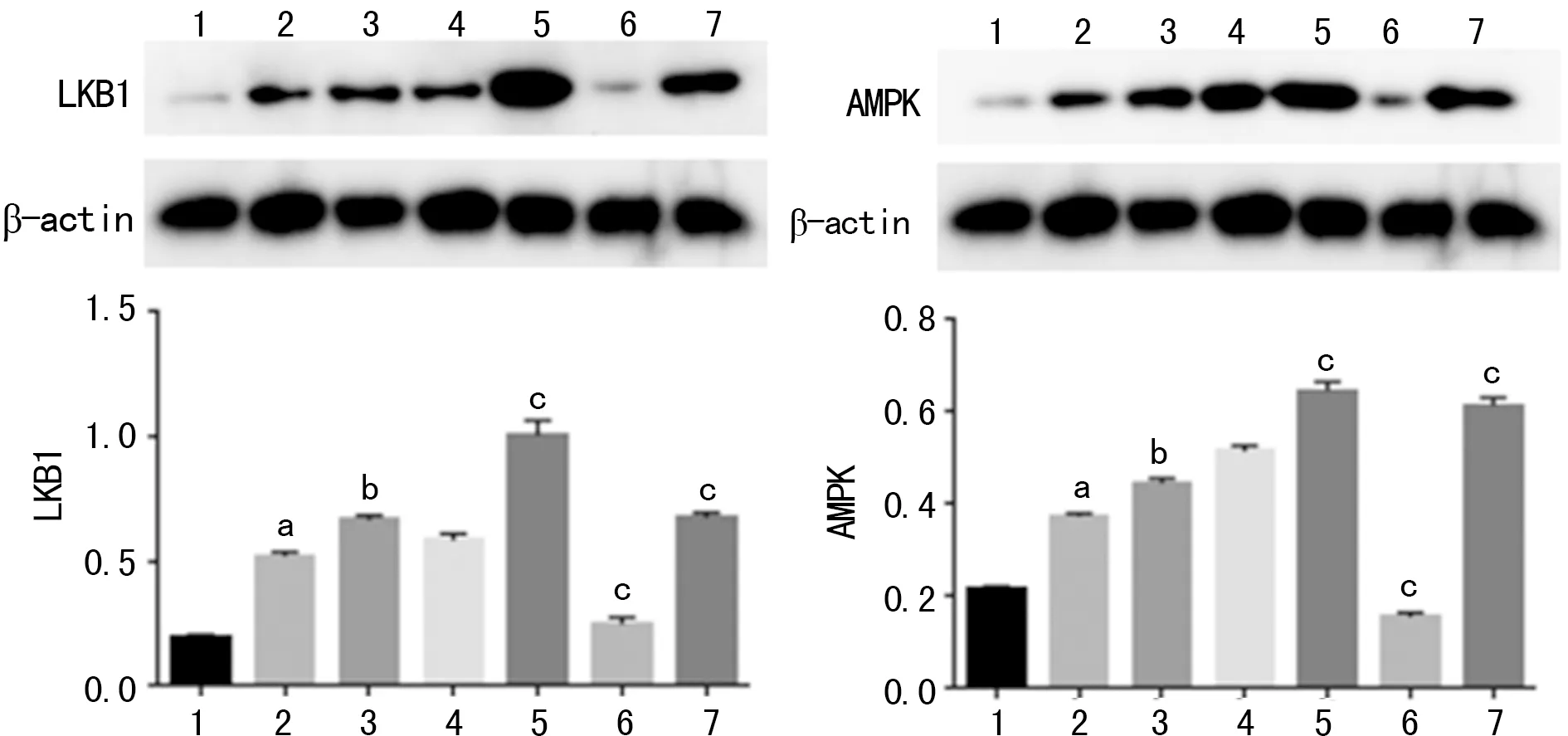
1:对照组;2:I/R组;3:I/R+HGF组;4:I/R+HGF+Ad-GFP组;5:I/R+HGF+GSK3β-Ad-wt组;6:I/R+HGF+GSK3β-Ad-S9A组;7:I/R+HGF+GSK3β-Ad-K85A组;a:P<0.05与对照组比较;b:P<0.05,与I/R组比较;c:P<0.05,与I/R+HGF组比较
图2 各组LKB1、AMPK表达情况

1:对照组;2:I/R组;3:I/R+HGF组;4:I/R+HGF+Ad-GFP组;5:I/R+HGF+GSK3β-Ad-wt组;6:I/R+HGF+GSK3β-Ad-S9A组;7:I/R+HGF+GSK3β-Ad-K85A组;a:P<0.05与对照组比较;b:P<0.05,与I/R组比较;c:P<0.05,与I/R+HGF组比较
图3 各组LC3Ⅱ、Beclin-1表达情况
2.4各组Akt表达情况 与对照组比较,I/R组的pAkt(Akt的Ser473位点磷酸化)的表达量明显升高(P<0.05)。与I/R组比较,I/R+HGF组pAkt明显升高(P<0.05)。而在HGF激活剂的作用下,各感染组的pAkt表达量差异有统计学意义(P<0.05),见图4。

1:对照组;2:I/R组;3:I/R+HGF组;4:I/R+HGF+Ad-GFP组;5:I/R+HGF+GSK3β-Ad-wt组;6:I/R+HGF+GSK3β-Ad-S9A组;7:I/R+HGF+GSK3β-Ad-K85A组;a:P<0.05与对照组比较;b:P<0.05,与I/R组比较;c:P<0.05,与I/R+HGF组比较
图4 各组Akt表达情况
3 讨 论
GSK3β是一种高度保守的丝氨酸/苏氨酸酶。GSK3β已被证明作用于多种信号蛋白和转录因子,调节细胞的分化、增殖、存活和凋亡[9]。既往研究表明,GSK3β在Ser9位点磷酸化从而失去活性在心肌缺血再灌注引起的炎性反应,钙超载和氧化应激反应等中具有抗炎和抗凋亡作用,从而保护心肌细胞,其机制可能与其提高线粒体的通透性转换孔(mPTP)开放阈值,减少线粒体损伤,细胞凋亡和坏死相关[6]。GSK3β作为HGF/Met信号通路中的一个下游信号蛋白,在HGF/Met的抗炎、抗凋亡等多种组织保护机制中起着关键性作用[7-8]。而GSK3β在心肌细胞发生缺血再灌注时HGF/Met信号通路促进自噬,保护心肌细胞中的作用,缺乏相关报道。
当心肌发生缺血再灌注时,除心肌细胞的坏死和凋亡程度影响预后,自噬的发生同样也影响心肌的梗死面积和存活[3]。其机制可能是当心肌发生缺血时,细胞的能量缺乏促进AMPK的激活,导致自噬基因Beclin-1的苏氨酸388位点产生磷酸化,促使自噬基因Beclin-1与Bcl-2解离,促进自噬相关蛋白Beclin-1和VPS34及Atg14的结合形成三聚体复合物,三聚体再不断地招募相关的核心蛋白,作为自噬起始;在自噬体的延长阶段,LC3Ⅰ会被包括Atg7和Atg3在内的泛素样体系所修饰和加工,产生相对分子质量为14×103的LC3Ⅱ,并定位到自噬小体中。这样,自噬小体中存在的LC3和低分子量的LC3Ⅱ都被当作细胞发生自噬的分子标志,并且LC3Ⅱ的含量和发生自噬的程度成正比。因此这两个蛋白可以作为自噬水平的检测指标[10]。本实验通过腺病毒转染大鼠离体心肌细胞的方法使GSK3β不同的位点发生突变从而使GSK3β处于持续失活或激活状态,并通过建立缺血再灌注模型,以GSK3β下游通路蛋白和自噬相关蛋白为指标研究在HGF激活剂的作用下,GSK3β的失活对促进细胞自噬,减轻细胞损伤的相关作用[11]。既往研究结果显示,在缺血状态下,HGF的激活可以激活下游PI3K/Akt/Bcl-2信号通路从而抑制细胞凋亡,保护心肌细胞[12]。GSK3β作为PI3K/Akt的下游信号蛋白在缺血时同样发挥着重要的作用。研究发现,抑制GSK3β的活性(GSK3β的Ser9位点磷酸化)能有效提高线粒体mPTP的开放阈值,减少线粒体损伤、细胞凋亡和坏死的发生[13]。本实验结果显示,与对照组相比,I/R组AMPK的表达量明显升高,验证了心肌细胞在发生缺血时会产生一定量的自噬。AMPK的激活与其上游基因LKB1的活性有关。在之前的报道中表明,GSK3β去磷酸化将会抑制LKB1的活性。本实验结果显示,HGF促进了pAkt、pGSK3β的表达,以及下游通路蛋白LKB1、AMPK和自噬相关蛋白Beclin-1、LC3Ⅱ的表达。证实了在缺血状态下,HGF对心肌细胞自噬的促进作用及GSK3β在HGF促进自噬中的可能性的潜在性作用。而在HGF激活剂的作用下,GSK3β的失活(Ad-wt和Ad-K85A腺病毒感染组)使下游通路蛋白LKB1、AMPK和自噬相关蛋白Beclin-1、LC3Ⅱ的表达量明显升高。表明了HGF/Met信号通路在心肌缺血再灌注损伤中通过抑制GSK3β的活性,促进细胞自噬到达保护作用。而持续激活GSK3β的Ad-S9A腺病毒感染组中自噬呈减弱趋势,可能导致心肌细胞加速损伤。
既往研究发现,HGF与其受体的结合可以激活下游信号蛋白PI3K/Akt,而PI3K/Akt的激活可以促进下游信号蛋白GSK3β在Ser9位点发生磷酸化,失去活性[6]。在心血管系统中,GSK3β在葡萄糖代谢、心肌肥厚和细胞死亡等病理过程中起着关键性作用[14]。在慢性脑缺血性疾病中,增加GSK3β的磷酸化会导致pmTOR的水平增加及Beclin-1水平的降低,从而抑制细胞破坏性自噬的活性。在对高糖血症和血脂异常的人主动脉内皮细胞的研究中发现,抑制GSK3β的活性可以通过增加AMPK的活性增加自噬体的形成,从而减少内皮细胞的死亡及动脉粥样硬化的形成[15]。结合本实验结果,表明GSK3β在HGF保护心肌对抗缺血再灌注损伤中起着重要的作用。本实验从蛋白水平证实了HGF通路的激活可以通过促进细胞自噬保护心肌细胞在缺血再灌注中的损伤,其可能机制是PI3K/Akt/GSK3β/AMPK信号通路的激活,但其是否存在其他可能的分子机制及作用靶点尚不明确,仍待进一步完善。
心肌细胞是一种高度分化的永久性细胞,其损伤往往是不可逆的,会引起致命性损伤。故减少心肌细胞的损伤和死亡对保护心脏极为重要。自噬在缺血再灌注中对心肌细胞起保护作用的同时也会引起细胞自噬性死亡,进一步加重心肌细胞的受损。因此,避免或尽最大程度地降低自噬在缺血再灌注损伤引起的细胞死亡将可能成为未来治疗冠心病的重要环节。
猜你喜欢
传染病信息(2022年3期)2022-07-15
油田化学(2020年1期)2020-04-07
天津医科大学学报(2019年6期)2019-08-13
灾害医学与救援(电子版)(2018年1期)2018-06-05
分析化学(2017年12期)2017-12-25
上海农业学报(2017年4期)2017-04-10
现代检验医学杂志(2016年1期)2016-11-12
罕少疾病杂志(2016年4期)2016-03-11
中西医结合心脑血管病杂志(2016年20期)2016-03-01
中国卫生标准管理(2015年6期)2016-01-14
