
载脂蛋白基因apo和gltP对普鲁兰多糖合成的影响
2019-04-22 09:27黄思瑶陈叶福郭学武肖冬光
天津科技大学学报 2019年2期
郭 建,黄思瑶,郑 鹏,陈叶福,郭学武,肖冬光
(工业发酵微生物教育部重点实验室,天津科技大学生物工程学院,天津 300457)
普鲁兰多糖是由出芽短梗霉合成的一种胞外多糖,它由麦芽三糖以α-(1→6)糖苷键反复连接而成[1].特殊的化学结构赋予普鲁兰多糖优良的性质,它黏度大、水溶性极强,在化工、食品、医药等行业应用广泛.普鲁兰多糖可作为增稠剂,也能用于生产可被微生物降解的薄膜,还可作为低卡路里食品直接食用[2].
普鲁兰多糖的合成是一个复杂过程.磷酸葡萄糖变位酶、葡萄糖焦磷酸化酶、葡萄糖基转移酶可能是普鲁兰多糖合成过程中的关键酶[1].Li等[3]过表达葡萄糖焦磷酸化酶基因后发现,普鲁兰多糖产量提升了1.3倍.Chen等[4]验证了几个葡萄糖基转移酶基因的功能,发现其中1个葡萄糖基转移酶基因对普鲁兰多糖产量有促进作用.以上实验结果同推测的普鲁兰多糖合成途径相互印证.除了上述关键酶外,糖脂中间体 lipid-G对多糖的合成也至关重要[5].糖脂中间体 lipid-G是由脂类物质与尿苷二磷酸葡萄糖(UDPG)结合而成的[5].已有的文献[3-4]报道都是从增加 UDPG含量的角度来增强普鲁兰多糖的合成,并未考虑脂类物质对糖脂中间体lipid-G的影响.
载脂蛋白(apolipoprotein)对脂类物质的合成和运输均有较大影响[5-6],脂类物质对于糖脂中间体的形成至关重要.脂类物质含量的增加能够促进糖脂中间体的形成,作为普鲁兰多糖合成的基本单元,足量的糖脂中间体无疑会促进普鲁兰多糖的合成.出芽短梗霉所产脂类物质的主要成分为重油[1],但是重油在普鲁兰多糖合成中的作用还未见报道,由此可以推测其可能参与糖脂中间体 lipid-G的合成.基于以上认识,本研究以出芽短梗霉(A.pullulans)CBS110374菌株为研究对象,通过敲除和整合过表达,考察了出芽短梗霉载脂蛋白基因 apo和 gltP对普鲁兰多糖及重油合成的影响,这有助于深入认识普鲁兰多糖合成过程.
1 材料与方法
1.1 菌株与试剂
出芽短梗霉(A.pullulans)CBS110374购自荷兰CBS菌种保藏中心,所用工程菌株 OA21、OG5、Δapo和ΔgltP为本实验构建.
DNA 聚合酶、dNTP,Takara公司;D9515崩溃酶、L4025消解酶、Trizma碱,Sigma公司;潮霉素B,上海索宝生物科技有限公司;山梨醇,上海迈瑞尔化学技术有限公司;甲醇,天津市北方天医化学试剂厂;无水乙醇,天津市申泰化学试剂有限公司.
1.2 培养基及培养条件
YEPD培养基(g/L):蛋白胨20,酵母粉10,葡萄糖 20,固体培养基中加入琼脂粉 15,121℃灭菌20min.
HCS培养基(g/L):葡萄糖 10,酵母浸粉 3,牛肉粉 1,蛋白胨 10,麦芽粉 3,固体培养基中加入琼脂粉 15,pH 5.7,115℃灭菌 20min.HCS培养基双层培养基的下层 HCS培养基添加终质量浓度为50mg/L的潮霉素,上层HCS培养基不添加潮霉素.
种子培养基(g/L):葡萄糖 20,硫酸镁 0.4,酵母粉 1.2,硫酸铵 1.0,NaCl 4,K2HPO44,pH 6.0,115℃灭菌20min.
发酵培养基(g/L):葡萄糖 50,硫酸镁 0.2,酵母粉 0.8,硫酸铵 0.8,NaCl 2,K2HPO46,pH 6.5,115℃灭菌20min.
种子培养条件为 28℃、200r/min培养 24h,发酵培养条件为28℃、220r/min培养7d.
1.3 重组菌株构建
重组菌株的构建采用同源重组依赖型 DNA装配方法[7],过表达gltP基因的菌株的构建过程参考文献[7].所用引物(表1)由金维智生物技术有限公司合成,下划线表示片段间重叠的碱基.

表1 引物Tab. 1 Primers

续 表
同源重组依赖型 DNA装配方法为相邻片段引物设计方法,在 rDNA 基因(Genbank NO.AY139394.1)位点整合过表达apo或gltP基因,如图1所示.前期研究[7]发现出芽短梗霉拥有高效的同源重组修复机制,基于这种机制在设计引物时于引物末端加入同源序列,然后将待整合片段导入细胞内,即可在细胞内完成片段连接并整合到设计好的位点.

图1 同源重组依赖型 DNA装配方法引物设计原则及整合过表达apo或gltP示意图Fig. 1 Principles of homologous recombination dependent DNA assembly method and the schema of integrating overexpression of apo or gltP
1.3.1 转化片段准备
在rDNA位点过表达apo基因(JGI protein ID:84794)需要 6个片段,即 HPT片段、rDNAU片段和rDNAD片段,PGKP5(磷酸甘油酸激酶基因启动子)用引物 PGKPF/PGKP5R扩增,apo片段(apo基因)用引物 apoU/apoD 扩增,PGKt5(磷酸甘油酸激酶基因终止子)用引物PGK5tF/PGKtR扩增.敲除apo基因需要 3个片段,apo基因上同源臂用引物ApoUs/ApoUa扩增,apo基因下同源臂用引物ApoDs/ApoDa扩增,潮霉素抗性基因用引物HPT3s/HPT3a扩增.
敲除 gltP基因需要 3个片段,gltP基因上同源臂用引物 gltPU1s/gltPU1a扩增,gltP基因下同源臂用引物 gltPD1s/gltPD1a扩增,潮霉素抗性基因用引物HPT4s/HPT4a扩增.
1.3.2 出芽短梗霉转化
出芽短梗霉采用电转化方法:取1环出芽短梗霉接种于5mL的YEPD试管中,28℃、200r/min培养24h;取 2mL菌液接种于装液量为 50mL YEPD的250mL 三角瓶中,28℃、200r/min培养 24h;2mL菌液移至 2mL灭菌离心管,4℃、12000r/min离心2min,收集菌体,可重复多次以获得足量菌体;用1mol/L山梨醇溶液将菌体重悬,4℃、12000r/min离心2min,收集菌体;加入1mL 溶液Ⅰ(1mol/L山梨醇和 50mmol/L柠檬酸钠混合液,pH 5.8)将菌体重悬,并加入适量的 D9515崩溃酶和 L4025消解酶(703U/mg)处理细胞,28℃、100r/min酶解 10min后迅速拿出,4℃、2500r/min离心 10min,收集菌体;菌体用 2mL溶液Ⅰ重悬,4℃、2500r/min离心10min,弃上清液,重复1次;菌体用2mL 1mol/L山梨醇溶液重悬,4℃、2500r/min离心 10min,弃上清液,重复 1次;菌体用100µL 1mol/L山梨醇溶液混匀,并加入预冷的待转片段,冰浴放置 10min;将混合液加入已经预冷好的电转杯中,冰浴放置 10min;1500V、200Ω 条件下进行电转,电转完毕后将菌体用1mol/L山梨醇溶液洗出,30℃培养箱中修复培养2~4h;修复培养后将全部菌体涂布于 HCS双层平板(初筛平板)中,30℃培养箱中培养 2~3d即可见转化子;将初筛平板长出的单菌落点板到 YEPD潮霉素抗性平板(复筛平板)上,30℃培养箱中培养 2d即可见转化子.
1.4 出芽短梗霉生物量和葡萄糖含量的测定
取 10mL菌液,12000r/min离心 2min,弃上清液;菌体用纯净水洗涤2次,12000r/min离心2min,弃上清液;所得菌体用纯净水悬浮后,转入干净的平皿中,70℃烘干至质量恒定,计算出芽短梗霉的生物量.
采用高效液相色谱法测定发酵液中葡萄糖含量:色谱柱为 Aglient HPX-87H(300×7.8mm),检测器为示差检测器(RID),检测器温度55℃,柱温65℃,流动相为5mmol/L硫酸溶液,流量0.6mL/min,进样量10µL.
1.5 普鲁兰多糖产量的测定
取 10mL菌液,12000r/min离心 2min取上清液 5mL,加入 2倍体积的无水乙醇,4℃放置 12h,5000r/min离心 8min去上清液;沉淀出来的多糖再用无水乙醇清洗2次;70℃烘箱烘干至质量恒定,计算多糖产量.
1.6 酶活力测定
依据 McCleary等[8]的方法测定葡萄糖基转移酶活力.将适当稀释的待测样本(0.2mL)与预均衡的对硝基苯基α-D-吡喃葡萄糖苷(0.2mL,10mmol/L)置于 0.1mol/L乙酸钠缓冲液(pH 4.0)中,40℃处理5min.通过加入 Trizma碱(3.0mL,2.0%)的水溶液终止反应,测量 410nm 处的吸光度.酶活力用对硝基苯酚释放量(µmol/L)表示,并通过参考标准曲线计算(通过用不同浓度的对硝基苯酚测量溶液的A410值来测定标准曲线).使用在 100℃加热 5min的样本和反应液的混合物作为对照.
根据 Dutra等[9]的方法测定 UDPG焦磷酸化酶活力.酶活力单位定义为在测定条件下在1min内催化 1.0µmol/L底物转化的酶量.使用在 100℃加热5min的样本和反应液的混合物作为对照.
根据Qian等[10]的方法测定磷酸葡萄糖变位酶活力.酶活力单位定义为在测定条件下在1min内催化1.0µmol/L底物转化的酶量.使用在 100℃加热5min的无细胞提取物的混合物作为对照.
1.7 重油含量的测定
取 20mL发酵液,加入等体积的氯仿-甲醇混合液(1∶1),在 50mL离心管中来回颠倒混匀;然后将混合液在4℃、12000r/min离心10min,将上清液倒入分液漏斗,待分层后将下层含有重油的液体分出;将含有重油的液体经氮吹仪氮吹,去除有机溶剂;得到的重油用氯仿-甲醇溶液再反复处理 2次,用以纯化重油;重油烘干至质量恒定,计算重油含量.
1.8 普鲁兰多糖结构的测定
普鲁兰多糖的结构采用傅里叶红外光谱仪进行分析.将 10mg样品与 250mg溴化钾混合均匀,用制片机在高压条件下制成薄片,仪器扫描波长范围为400~4000cm-1.
1.9 mRNA水平的测定
mRNA水平的测定方法参见文献[7].测定 apo基因用引物 APOs/APOa,测定 gltP基因用引物gltPa/gltPs.
2 结果与分析
2.1 apo和gltP对出芽短梗霉菌株生长性能的影响
通过 NCBI查询到出芽短梗霉包含两个载脂蛋白基因 apo和 gltP,分别构建了整合过表达 apo或gltP基因的菌株 OA21和 OG5,以及单敲 apo或者gltP基因的菌株 Δapo和 ΔgltP.重组菌株的生长情况和葡萄糖利用能力如图 2所示.敲除 apo或 gltP后,菌株生长受到抑制,生物量分别较亲本株减少了10.85%和16.84%,而整合过表达apo或gltP对生物量没有显著影响.敲除 apo或 gltP降低了菌株对葡萄糖的利用能力,发酵 7d消耗的葡萄糖分别较亲本株减少了 4.5%和 13.3%,而整合过表达 apo或 gltP增强了菌株的葡萄糖利用能力,发酵 6d后整合过表达apo或gltP基因的菌株已经将葡萄糖消耗殆尽.

图2 重组菌株生物量和葡萄糖利用能力同亲本株的比较Fig. 2 Comparison of the cell growth and glucose utilization ability
2.2 apo和gltP对普鲁兰多糖合成的影响
为了考察载脂蛋白基因apo和gltP与普鲁兰多糖合成的关系,对所得重组菌株的普鲁兰多糖合成能力进行了测定,如图3所示.整合过表达apo或gltP后,普鲁兰多糖产量分别较亲本株增加了 7.78%和15.23%.敲除apo或gltP抑制了普鲁兰多糖的合成,普鲁兰多糖产量分别较亲本株降低了 21.35%和35.07%.由此可知,相较于 apo基因,gltP基因对于普鲁兰多糖的合成更为重要.
2.3 普鲁兰多糖合成途径关键酶活力
为了深入探究apo和gltP与普鲁兰多糖合成的关系,对普鲁兰多糖合成途径中的3个关键酶的酶活力进行了测定,结果见表 2.由表 2可知:整合过表达 apo后,磷酸葡萄糖变位酶活力、UDPG焦磷酸化酶活力和葡萄糖基转移酶活力分别较亲本株增加了18.52%、16.60%和 15.79%.整合过表达 gltP后,磷酸葡萄糖变位酶活力、UDPG焦磷酸化酶活力和葡萄糖基转移酶活力分别较亲本株增加了 25.93%、30.23%和 36.84%.敲除apo或gltP后,磷酸葡萄糖变位酶活力、UDPG焦磷酸化酶活力和葡萄糖基转移酶活力均出现不同程度的降低.

图3 apo和gltP对普鲁兰多糖合成的影响Fig. 3 Effect of apo and gltP genes on the synthesis of pullulan

表2 重组菌株普鲁兰多糖合成途径关键酶活力Tab. 2 Enzyme activity of the key enzymes in the pullulan synthesis pathway
2.4 apo和gltP对以重油为代表的脂类物质合成的影响
载脂蛋白与脂类物质的合成密切相关.出芽短梗霉所产脂类的主要成分为重油[1].为了考察载脂蛋白基因 apo和 gltP与脂类物质合成之间的关系,选取重油作为脂类物质的代表进行研究.出芽短梗霉分泌的重油是一种脂类物质,在医药、化工等行业有巨大的应用前景,apo和gltP对以重油为代表的脂类物质合成的影响如图4所示.由图4可知:整合过表达 apo或 gltP后,重油产量分别较亲本株增加了41.38%和3.5倍.敲除apo或gltP抑制了脂类物质的合成,重油产量分别较亲本株降低了 17.24%和28.74%.这些结果表明,apo和gltP都能影响出芽短梗霉对重油的合成,但是gltP基因的影响更显著.

图4 apo和gltP对重油合成的影响Fig. 4 Effect of apo and gltP genes on the synthesis of heavy oil
2.5 普鲁兰多糖结构鉴定
采用傅里叶红外光谱仪对重组菌株所产普鲁兰多糖结构进行了鉴定,并同普鲁兰多糖标品进行比对,结果如图 5所示.由图 5可知:所有样品均在755cm-1和 915cm-1处有吸收峰,这些特征峰证实了α-(1→4)和α-(1→6)糖苷键的存在,而这个键型结构是构成普鲁兰多糖最基本的结构.同普鲁兰多糖标品的红外光谱图比对发现,所有重组菌株与标品结构基本一致,确认了所产胞外多糖为普鲁兰多糖.

图5 重组菌株所产普鲁兰多糖与普鲁兰多糖标品红外光谱的比对Fig. 5 Comparison of the IR spectra of pullulan produced by the recombinant strains with that of the standard pullulan
2.6 获得的重组菌株apo和gltP基因相对表达量
对敲除或整合过表达 apo,以及敲除或整合过表达 gltP基因的菌株所对应基因的转录水平进行了检测,见图 6.从图 6中可知,敲除 apo或 gltP基因后apo或 gltP基因转录几乎检测不到,进一步证实了apo或gltP基因已经被敲除.整合过表达apo或gltP则显著增强了对应基因的转录水平,OA21菌株的apo转录水平较原始菌株增加了4.1倍,OG5菌株的gltP转录水平较原始菌株增加了3.9倍.
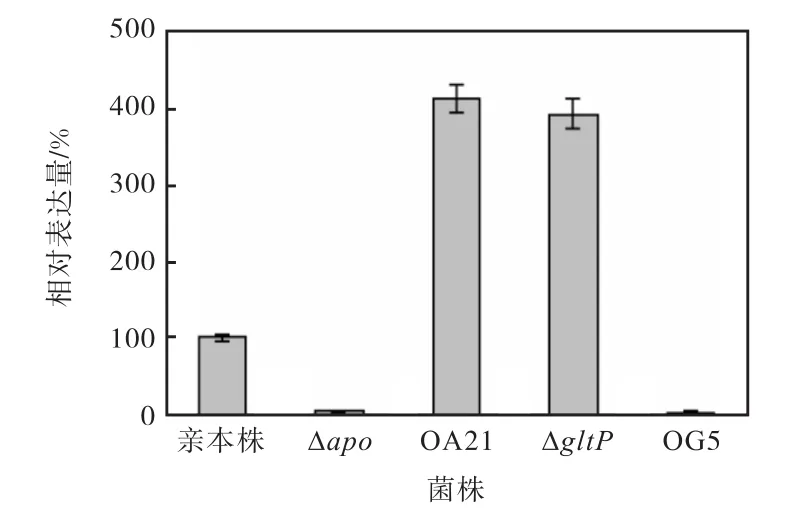
图6 重组菌株对应基因转录水平与亲本株的比较Fig. 6 Comparison of the transcription levels of the recombinant strains with parent strains
3 讨 论
普鲁兰多糖理化性能独特,在食品、化工、医药等行业应用广泛[1].经文献汇总,本文绘制普鲁兰多糖合成途径(图7).

图7 普鲁兰多糖合成途径Fig. 7 The proposed pullulan synthesis pathway
已有的文献报道部分印证了该合成途径,葡萄糖基转移酶、磷酸葡萄糖变位酶、UDPG焦磷酸化酶和普鲁兰合酶被认为是普鲁兰多糖合成途径中的关键酶[1,3-4].除了上述关键酶外,脂类物质也同普鲁兰多糖合成密不可分.脂类物质与 UDPG的结合形成普鲁兰多糖合成的基本单元lipid-G[5],而这是普鲁兰多糖合成所必须的.载脂蛋白负责调控脂类物质的生物合成[6],那么载脂蛋白势必会影响普鲁兰多糖的合成.多位研究者[5-6,11]也认为胞外多糖的合成与脂类物质紧密相关.
为了确认脂类物质是否参与普鲁兰多糖的合成过程,本文考察了载脂蛋白基因 apo和 gltP对普鲁兰多糖合成的影响.载脂蛋白基因 apo和 gltP能够影响出芽短梗霉生长性能和葡萄糖利用能力.apo或者 gltP的敲除抑制了菌株的生长,这可能是由载脂蛋白对胞内多种细胞器膜的调控引起的[12].敲除apo或者 gltP后菌株葡萄糖利用能力降低,这是由较慢的菌株生长和生成产物的减少引起的(图3和图4).
载脂蛋白能调节脂类物质的合成和运输[13].本文研究结果表明整合过表达apo或者gltP能够促进以重油为代表的脂类物质和普鲁兰多糖的合成,而敲除 apo或者 gltP则抑制了重油和普鲁兰多糖的生成.由此可知,适量增强出芽短梗霉脂类物质的合成有助于提高普鲁兰多糖产量,这与 Sutherland[11]和Simon等[6]的推测是相符合的.Price等[14]的研究表明,过度强化出芽短梗霉脂类物质的合成不利于普鲁兰多糖合成.研究结果还发现,虽然apo和gltP能够调控普鲁兰多糖的产量,但是并不引起普鲁兰多糖结构的改变.
通过测定重组菌株普鲁兰多糖合成途径中关键酶的酶活力,发现整合过表达 apo或者 gltP能够促进葡萄糖基转移酶、磷酸葡萄糖变位酶和 UDPG焦磷酸化酶的活力,而敲除 apo或者 gltP则降低了 3个关键酶的活性.葡萄糖基转移酶、磷酸葡萄糖变位酶和 UDPG焦磷酸化酶对普鲁兰多糖合成至关重要[1,3-4],这3个关键酶活力的改变能直接影响普鲁兰多糖的合成.
重油是普鲁兰多糖所产脂类物质的主要成分.研究结果显示普鲁兰多糖产量和重油产量呈正相关,这预示着重油可能参与普鲁兰多糖合成所需基本单元糖脂中间体 lipid-G的生成.但是,并非重油合成能力越强普鲁兰多糖合成能力越强,比如 Price等[14]就发现过度强化的重油合成能力能够抑制普鲁兰多糖的生成.
apo和 gltP对出芽短梗霉普鲁兰多糖合成的调节作用可以从以下几点来解释.第一,apo和 gltP调控脂类物质的合成,进而影响普鲁兰多糖合成所需基本单元糖脂中间体 lipid-G的生成,最终改变普鲁兰多糖合成.第二,apo和 gltP表达量的改变引起了脂类物质含量的改变,某些脂类物质作为反式作用因子强化了 UDPG 的合成[15],导致普鲁兰多糖合成途径中关键酶活力性的增加.第三,出芽短梗霉重油是一种表面活性剂[16],重油合成的增强有助于改善出芽短梗霉细胞膜通透性[17],有利于普鲁兰多糖的外泌.
综上,本文研究结果表明载脂蛋白基因 apo和gltP能够调节出芽短梗霉普鲁兰多糖的合成,并且gltP对普鲁兰多糖合成的影响大于 apo.gltP和 apo都负责介导脂类物质的转运,但 gltP主要负责脂类物质向胞外转运[7],而 apo负责胞内转运[18],不同的分工导致了这两个基因对于脂类物质合成的作用相区别.
4 结 语
本文研究结果显示,整合过表达 apo或 gltP能使普鲁兰多糖产量分别增加 7.78%和 15.23%,而分别敲除apo或gltP则使普鲁兰多糖产量降低21.35%和 35.07%;证实了脂类物质和普鲁兰多糖合成紧密相关,丰富和完善了普鲁兰多糖合成途径,为增加普鲁兰多糖合成基本单元糖脂中间体lipid-G的来源提供了新的思路.
猜你喜欢
化学工业与工程(2022年1期)2022-03-29
能源化工(2021年6期)2021-12-30
能源工程(2021年1期)2021-04-13
中国甜菜糖业(2020年1期)2020-12-07
家庭百事通·健康一点通(2020年11期)2020-11-30
青岛大学学报(医学版)(2020年6期)2020-11-16
化工设计通讯(2020年6期)2020-06-20
船舶标准化工程师(2020年1期)2020-06-12
江苏农业科学(2016年1期)2017-05-17
家庭医学(2015年6期)2015-07-03
