
CRISPR/dCas9在细菌转录调控中的应用及展望
2019-04-22 09:27:18黄龙辉刘其敬谢燕燕谭之磊
天津科技大学学报 2019年2期
钟 成,黄龙辉,刘其敬,谢燕燕,谭之磊
(工业发酵微生物教育部重点实验室,天津科技大学生物工程学院,天津 300457)
CRISPR(clustered regularly interspaced short palindromic repeats)是细菌等生物体细胞内的一种遗传适应性免疫系统,细菌可以通过该系统抵御侵入细胞内的 DNA[1].它主要由 crRNA(CRISPR RNA)和相应的 Cas(CRISPR-associated)蛋白组成.在 CRISPR系统中,crRNA的基因包含一段用于定位 CRISPRCas系统靶位点基因的序列,该序列是一段外源侵入至 CRISPR阵列的 DNA.crRNA主要通过结合 Cas蛋白和特异的 20nt左右的间隔序列对 DNA序列进行特异的靶边点识别,并实现对靶位点 DNA的切割[2-3].通过整合外源侵入的 DNA,CRISPR 能够记录下外源 DNA的具体序列,从而实现对外源 DNA特定序列的切割.基于不同 CRISPR免疫系统中crRNA和Cas蛋白的不同,CRISPR-Cas系统主要通过其特征基因分为三大类型:Ⅰ型系统具备 cas3,Ⅱ型系统具备 cas9,Ⅲ型系统具备 cas10.在所有的CRISPR/Cas系统中,Ⅱ型系统的结构最为简单,仅需要 Cas9蛋白便能实现切割,因此人们最早将它改造成用于基因编辑的CRISPR/Cas9系统[4].
CRISPR/dCas9是通过将 CRISPR/Cas9系统中的Cas9蛋白改造成dCas9(catalytically dead Cas9),得到的具有转录调控功能的系统.相对于 CRISPR/Cas9系统,CRISPR/dCas9系统具备与靶位点结合,但不对DNA进行切割的特点(图1).
Cas9蛋白的结构如图2所示,它主要存在HNH和RuvC核酸酶结构域以及与sgRNA(target-specific guide RNA)接触的α-螺旋叶,其中HNH和RuvC核酸酶结构域都是切割 dsDNA所必需的[5-6].HNH和RuvC两个结构域各自负责一条单链的切割,选择其中任何一个结构域进行突变可以得到具备单链切割能力的 Cas9n蛋白.将两个不同突变位点的 Cas9n蛋白结合起来切割双链,从而提高靶位点切割的特异性[7].将 CRISPR系统中 Cas9蛋白改造成 dCas9蛋白后,蛋白就能方便精准地被引导至靶位点.同时,对HNH和RuvC两个结构域中的D10A和H840A氨基酸进行点突变可以获得剪切失活的Cas9蛋白[8].基于此,研究人员除了将dCas9蛋白直接用于转录抑制外,也融合了不同的转录调控相关的因子,得到了一系列基于CRISPR/dCas9的转录调控工具.
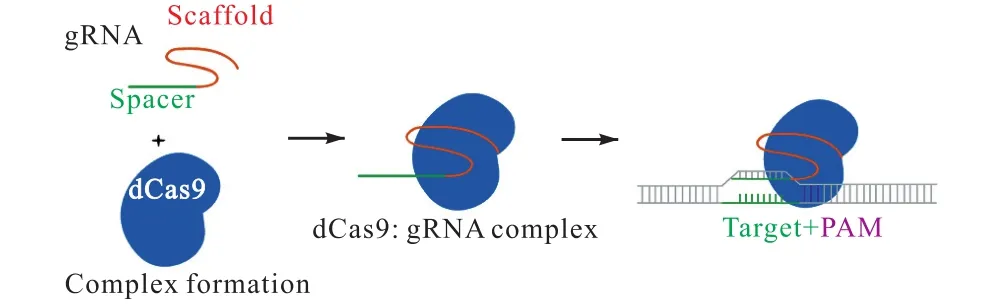
图1 Cas9蛋白与gRNA及靶位点的结合过程Fig. 1 The binding process of Cas9 to gRNA and target sites

图2 Cas9蛋白的结构示意图Fig. 2 Schema of Cas9 protein structure
Cas9首先需要识别一个短的原型间隔区相邻基序(protospacer adjacent motif,PAM),再通过 sgRNA中和 DNA互补的 20nt与 DNA结合.结合后的Cas9-sgRNA复合物就变得非常稳定;体外结合动力学研究结果表明,这一反应在结合后较长时间内几乎是不可逆的[9].通过对 CRISPR系统的改造,其已经在激活或抑制特定靶位点基因表达以及表观修饰等领域取得了重大突破[10-11].CRISPR/Cas9系统作为一种基因编辑工具,为基因工程带来了巨大的变革,许多综述已经详细阐述了其在真核生物中的应用[12-16].相比 CRSPR/Cas9系统,CRSPR/dCas9系统是在其基础上发展而来的转录调控工具.该系统作为转录抑制工具方面已在多种细菌中得到应用,而该系统在细菌作为转录激活工具的应用却鲜有报道.本文以 CRSPR/dCas9为例,首先对 CRISPR干扰和激活在细菌中的应用和研究进展进行了介绍,然后总结和讨论了在细菌中构建该系统存在的问题.
1 CRISPR干扰
细菌 CRISPR 干扰(CRISPR interference,CRISPRi)在大肠杆菌(E.coli)中已有大量研究[10-11,17].在大肠杆菌中,dCas9-sgRNA复合物通过阻断 RNA聚合酶(RNA polymerase,RNAP)与启动子 DNA 的结合或通过阻断转录的延伸抑制转录(图 3),这一点已被深度测序证明[10-11,18].靶向非模板链的延伸区段是最有效的[19].RNA测序表明这种抑制具有高度的特异性[10].CRISPRi已被用于模式菌株如大肠杆菌中的遗传回路控制[10]以及结核分枝杆菌(Mycobacterium tuberculosis)中基因表达的调控[20],并通过 CRISPRi改变代谢通量,从而改变代谢途径[21].研究[22]显示,CRISPRi系统已成功应用于革兰氏阳性细菌枯草芽胞杆菌(Bacillus subtilis)、人类微生物群共生菌拟杆菌(Bacterioides sp.)以及产生医学相关次级代谢产物的放线菌目中.

图3 CRISPRi系统的作用机制Fig. 3 Mechanism of the CRISPRi System
在细菌中dCas9仅需通过gRNA的引导与靶位点结合就能有效地抑制基因的表达.而与原核生物相比,在真核生物中dCas9蛋白不能明显地抑制基因表达,Cas9蛋白需要与转录抑制蛋白结构域(KRAB)融合,实现抑制基因的转录[23](图 3).与传统的调控基因表达的技术相比,CRISPRi的优势明显[24-26].第一,仅通过在sgRNA中插入一个新的20nt碱基配对区域就可以很容易地抑制新靶点.第二,由于CRISPRi构建完成后只需要改变其中的 20nt就可以靶向不同的基因,因此可以使用合并克隆策略,将20nt文库与CRISPRi载体整合进行文库构建[24,27-29].通过文库的构建,可以高通量地筛选出理想表型的菌株,并找到影响该表型的基因,从而进一步指导理性地改造菌株.第三,由于 CRISPRi可以是诱导型的,所以它能够通过部分抑制而操纵基因表达[20].
除了传统的诱导方法外,近年来有研究显示可以通过光遗传工具调控CRISPRi的表达.例如,通过光诱导 CRISPRi的表达调控哺乳动物细胞中基因的表达[30-31].而这一策略在发酵领域的应用虽未报道,但其在特定的发酵条件(比如静息发酵)下的基因调控同样具有相当价值.CRISPRi可与多种 sgRNA在同一细胞中同时对多种基因的表达进行干扰[10,17].这对于需要调节许多基因表达优化代谢途径的应用很重要,它可以简单快速地探究多个基因的表达对代谢通路的影响.由于这一系统构建方便,而在一些倍增时间极长的细菌中进行基因编辑又非常耗时,因此通过这种方法探究代谢途径的应用价值具有明显的优势.
然而,由于 dCas9阻断了 RNA聚合酶的延伸,操纵子中的下游基因的表达也同样会受到干扰,相对于基因组编辑来说,这是一个不可忽视的缺陷.与CRISPR编辑的情况一样,在CRISPRi对生长所必需基因进行干扰的时候,生存选择压力可能导致CRISPR系统发生突变甚至不再工作.最近,Cui等[19]将包含92000种特异性靶点的CRISPRi文库应用于大肠杆菌中高通量筛选时发现,sgRNA中靠近 PAM序列的12nt的种子序列中的5个特定的核苷酸就能造成菌株产生适应性缺陷.同时,他们也发现随着dCas9浓度降低,适应性缺陷也会降低.而 dCas9在低浓度时也能够明显抑制靶点基因的表达.因此,在构建细菌的CRISPRi过程中优化dCas9的表达量就显得尤为重要.
2 CRISPR激活
CRISPRa(CRISPR activition)是一种基于CRISPR/dCas9的转录激活工具.CRISPR/dCas9系统要实现转录激活,需要dCas9蛋白与转录激活因子融合或者在 sgRNA的末端融合一段 scRNA(small cytoplasmic RNA)来募集能激活基因表达的RNA结合蛋白,从而激活或增强基因转录.大肠杆菌CRISPRa系统中,dCas9与 RNA聚合酶的 ω亚基(dCas9∷ω)融合[11],在特异性的 sgRNA 引导下,RNA聚合酶被聚集到启动子的上游位置,从而激活转录[14](图 4).真核微生物中有 SAM、SunTag和VP64-p65-Rta(VPR)等系统[21,24-25].基于对不同启动子的特征分析表明,可能存在有效激活的狭窄区域,即 dCas9∷ω过于接近启动子则发生抑制,并且当dCas9∷ω的结合位点远离启动子时,激活则会失败.这一特性虽然使 CRISPRa不能明确界定靶位点,但同时也增强了 CRISPRa系统的特异性.CRISPRa对弱启动子的激活有明显效果[11].由于基因组中启动子的具体位点不能明确界定,因此该系统需要进一步的研究和优化才能广泛适用.研究[11]显示,大肠杆菌CRISPRa系统需要在缺乏ω因子的菌株中才能工作.
相对于传统的转录调控工具,CRISPRa系统继承了 CRISPR/dCas9系统在构建方面的简单快捷的优势.另外,其在利用转录调控探究基因功能方面能够弥补 CRISPRi对表达弱的基因不明显这一缺陷.不同基因表达的强弱不同,当 CRISPRa用于强启动子的时候其效果就不十分显著,而当 CRISPRi用于弱启动子的时候其效果也可能会不明显.因此,若利用 CRISPRa探究加强表达基因的功能或用CRISPRa探究削弱表达基因的功能,效果则不会显著.与此相反,当 CRISPRa用于弱启动子或CRISPRi用于强启动子的时候其效果则可能变得显著.因此,将二者分别用于同一基因的激活和弱化的时候,就可以抵消之前二者的缺陷.设计不同的 20nt序列和scRNA序列可以实现分别对不同基因的抑制和激活,并且两个系统互不干扰[34].结合 CRISPRi和CRISPRa就可以在基因组规模上进行基因功能的探究[32](图 5).CRISPRa系统的另一个应用是探究抗生素作用机制.为了实现这一效果,可能需要不依赖于启动子的精确间距的 CRISPRa的策略.研究人员通过在 sgRNA的末端连接长而灵活的连接子(一段能够与激活子结合的 RNA片段),利用连接子将激活子聚集在 dCas9附近[24,28](图 4),以减少CRISPRa对间距的依赖性.

图5 同时激活和抑制基因组上不同基因Fig. 5 Simultaneous activation and depression of different genes on a genome scale
3 应 用
由于细菌的多样性,CRISPR技术在细菌中的应用却不如哺乳动物中那么成熟[33].CRISPR系统在 γ变形菌门中基因编辑的实现标志着在原核微生物中从传统基因编辑技术到利用 CRISPR系统对基因组编辑的转变[34-35].厚壁菌和放线菌中的应用标志着研究人员正将这一便捷的技术转移到以前缺乏足够遗传改造工具的细菌中[36-37].将CRISPR/dCas9系统应用到一株新的菌株中主要会经历系统的选择与构建、靶位点的选择和gRNA的设计3个过程,本节将结合笔者实际研究和文献总结,从这 3个方面探讨CRISPR/dCas9在细菌转录调控中的应用以及应用过程中存在的问题.
(1) 利用 CRISPR/dCas9系统在一株新的细菌中有效地调控基因的表达需要选择合适的系统.而在系统选择的过程中,靶点基因表达的强弱是选择系统的主要依据.这是因为用该系统去激活表达强的基因或者弱化表达弱的基因都不会有明显的效果.因此,利用 CRISPR/dCas9探究细菌中某一基因功能的时候,将激活和弱化二者结合运用或许会取得更明显的效果.
(2) 需要考虑CRISPR/dCas9的表达系统.一方面,由于原核微生物的多样性,很难找到一个能在所有细菌中共用的表达系统,因此要将CRISPR系统引入一株新的菌株,需要选择能在宿主中工作的表达系统.另一方面,则是要在较低的水平表达 dCas9蛋白,这是因为过量的表达dCas9蛋白会对宿主造成毒害.而将dcas9基因的表达量控制在较低的表达水平则会有效地减少 CRISPR/dCas9系统对宿主的毒性[19]. 并且,Cas9-sgRNA复合物与基因结合后就变得非常稳定;体外结合动力学研究结果表明,这一反应在结合后较长时间内几乎是不可逆的[9].总而言之,在一个新的菌株中构建 CRISPR/dCas9系统时,需要考虑质粒的拷贝数、启动子的强弱和密码子的适应度的优化,让该系统达到既能对靶位点基因有效地调控又不至于对宿主带来毒性的效果.
(3) 需要选择合适的靶位点.与 CRISPR/Cas9这一基因编辑工具只需要考虑 Cas9-sgRNA复合物与靶位点的结合效率不同的是,在选取 CRISPR/dCas9靶位点的时候还需要考虑复合物结合靶位点后对基因调控的效果.在同一操纵子内,选取不同的靶位点具有不同的调控效果.就CRISPRi而言,可以选与结构基因启动子或基因内部结合.当在靶基因内部选取靶位点时,只有以编码链中的序列为靶位点时才能抑制基因的表达.而当将靶向位点选取在启动子的时候,以 DNA双链的任何一条链作为靶位点都会对基因的表达产生较强的抑制.当操纵子中有多个基因时,靶向上游基因能抑制下游基因的表达,而靶向下游基因对上游基因的表达几乎没有影响[19].与 CRISPRi靶位点选取不同的是,CRISPRa需要将靶位点选在启动子上游位置才能激活基因的表达[11].由于 CRISPRa可能存在狭窄的有效激活域,因此选取靶位点的时候最好不要太靠近启动子,也不要距离启动子太远.
(4) 设计有效的 sgRNA.sgRNA 的设计必须符合两个标准:靶向具有 PAM 的区域和最小化的脱靶率.选择靶向PAM序列是由于CRISPR虽然能灵活且精确地将 Cas9靶向基因组上不同位置,但靶向DNA特异位点首先需要Cas9识别PAM序列[8-9],然后通过 20nt左右的靶序列与 sgRNA的间隔区碱基配对.选择与PAM相邻靶位点完全匹配的20nt序列能有效降低脱靶率.这是因为虽然不完全匹配的靶位点仍然可以与 dCas9-sgRNA复合物结合,但抑制效果最强的 sgRNA都与 PAM 相邻靶位点完全匹配.在sgRNA的3′末端或与PAM序列相邻的12个核苷酸中的单个错配会明显降低 dCas9-sgRNA对基因表达的抑制作用.sgRNA与靶点之间的错配将进一步降低抑制的效果[10-11].抑制效果也可以通过改变靶点匹配所在基因内的位置来调整,这是由于目的基因的3′碱基或sgRNA与模板链结合的位置都会影响抑制效果[10,20].Bowtie等计算工具可用于比较PAM 相邻的靶位点,使用加权算法可以帮助丢弃具有潜在脱靶效应的sgRNA[29-30].
4 展 望
相对于在哺乳动物中CRISPRi需要与dCas9融合染色质沉默子(KRAB)这一特点[29,38],在细菌中CRISPRi系统仅 dCas9就能对基因的表达起到明显的抑制效果[10].因此,细菌 CRISPRi中 sgRNA的设计规则可能与哺乳动物细胞中的规则不同;这些规则可以在包含大量的 sgRNA文库实验中,利用深度测序测试sgRNA的功效进行确定.由于CRISPRi对基因的抑制会影响操纵子下游基因的表达,而不是仅对个别基因的抑制,因此 CRISPRi只适用于操纵子的筛选.另外,高浓度表达 dCas9对细菌造成适应性缺陷,因此在利用 CRISPR/dCas9系统探究细菌基因功能的时候,优化dCas9的表达量就显得尤为重要.
在细菌中构建 CRISPRi系统的过程中,还必须解决 CRISPRi系统在不同细菌中通用性的问题.由于不同细菌之间的种间差异较大,因此很有可能不会有通用的 CRISPRi系统能在所有细菌中工作.而将系统中的组件模块化标准化方法(比如可以通过酶切方法轻易地替换系统中的复制子或启动子等原件)或许更适用于CRISPR系统在不同细菌中的扩展.
与 RNAi或其他已知的调控基因表达的方法(基因过表达,TALE或ZF介导的调控)相比,CRISPRi/a具备设计简易性和高特异性的特点[23-24,38].基于这些特点,研究人员一方面可以通过其直接控制内源基因转录水平上的表达.另一方面也可以在该系统的基础上进一步改造,以适应不同的研究目的.比如,在基因互作方面,科研人员将 CRISPR/dCas9系统与荧光蛋白结合用于基因定位,研究活细胞中基因组的三维结构和动力学特征[7,10];在调控基因表达方面,研究人员将光遗传工具与 CRISPR/dCas9结合,实现了通过光诱导抑制基因的表达[26,32].而这些也为在细菌中进一步研究 CRISPR/dCas9提供了思路.CRISPR/dCas9技术以调控基因表达开始出现在实验室,相信其还有更多的扩展功能等待着研究人员去开发.
猜你喜欢
生物技术通报(2023年2期)2023-03-07 12:54:58
保健医苑(2022年5期)2022-06-10 07:46:38
中国临床医学影像杂志(2021年6期)2021-08-14 02:21:56
肝博士(2020年5期)2021-01-18 02:50:18
学苑创造·A版(2020年12期)2020-01-07 14:07:23
中国外汇(2019年15期)2019-10-14 01:00:34
作文教学研究(2016年1期)2016-07-05 12:22:47
医学研究杂志(2015年8期)2015-06-22 14:00:57
医学研究杂志(2015年7期)2015-06-22 11:01:01
中国药业(2014年21期)2014-05-26 08:56:45
