
线粒体自噬紊乱在糖尿病肾病发病机制中的作用研究进展
2019-04-09 12:34:34易鑫尧郭婷莉颜文慧陈莉娜
中国药理学与毒理学杂志 2019年11期
易鑫尧,郭婷莉,张 萌,郁 叶,颜文慧,陈莉娜
(西安交通大学医学部基础医学院药理学系,陕西 西安 710061)
自噬是依赖于溶酶体的细胞降解过程,对维持细胞结构稳定和功能的平衡至关重要,自噬紊乱和人类许多疾病包括癌症、神经退行性疾病以及糖尿病肾病(diabetic nephropathy,DN)等密切相关。自噬可分为巨自噬、微自噬和分子伴侣介导自噬[1]。线粒体自噬属于巨自噬,通过自噬途径将受损的线粒体清除以维持细胞内的稳态。根据自噬受体的分类可将哺乳动物中的线粒体自噬大致分为3类,分别是以P62、BRCA1邻近基因1蛋白(neighbor of BRCA1 gene 1,NBR1)和视神经蛋白(optineurin,OPTN)为自噬受体的PTEN诱导激酶1(PTEN-induced putative kinase1,PINK1)/帕金森病蛋白2(parkinson protein2,parkin)介导的线粒体自噬、以Bcl2/腺病毒E1B相互作用蛋白(recombinant Bcl2/adenovirus E1B 19 ku interacting protein,BNIP)为自噬受体的线粒体自噬以及以FUN14结构域包含蛋白1(FUN14 domain-containing 1,FUNDC1)为自噬受体的线粒体自噬[2]。哺乳动物中主要以PINK1/Parkin介导的线粒体自噬为主,其步骤包括自噬前体的形成、自噬体的形成、自噬溶酶体的形成以及线粒体的降解,详细过程分述如下:①线粒体自噬信号引发自噬激活激酶1(unc-51 like autophagy activating kinase 1,ULK1)复合物形成,ULK1复合物激活Ⅲ型磷脂酰肌醇3激酶(classⅢphosphatidylinositol 3-kinase,PI3KC3)/Vps34复合物并产生3-磷酸磷脂酰肌醇〔phosphatidylinositol 3-phosphate,PI3P〕,PI3P标记一个特定的内质网形成自噬前体[3]并招募效应蛋白WD重复结构域磷酸肌醇相互作用蛋白2(WD repeat domain phosphoinositideinteracting protein 2,WIPI2)和双重FYVE结构域包含蛋白 1(double FYVE-containing protein 1,DFCP1)[4];② 微管相关蛋白轻链 3-Ⅰ(microtubule-associated protein light chain 3-Ⅰ,LC3-Ⅰ)[5]与磷脂酰乙醇胺(phosphatidyl ethanolamine,PE)结合生成LC3-Ⅱ并通过WIPI2和DFCP1定位于吞噬泡上[6]。线粒体膜电位去极化使PINK1(PTEN-induced putative kinase 1,PINK1)在线粒体外膜聚集并从胞浆中招募Parkin。Parkin将大量的线粒体外膜蛋白泛素化,使自噬受体定位于线粒体外膜[7-9]。自噬受体通过其特异性的LC3相互作用序列(LC3-interacting region,LIR)与LC3相结合并将线粒体转移到吞噬泡中,吞噬泡不断延伸最终封闭形成自噬体;③自噬体和内溶酶体融合形成自噬溶酶体,然后自噬溶酶体中的酸性水解酶将线粒体降解从而将损坏的线粒体清除。
糖尿病是全球常见的一种慢性内分泌代谢疾病,可引发多种并发症。糖尿病血管病是最严重的并发症之一,其在临床上表现为微血管和大血管并发症。DN属于微血管并发症,它能引起40%~50%的终期肾疾病[10],并且是糖尿病患者的主要死亡原因之一。DN的发病机制十分复杂,包括糖基化终产物的增多、氧化应激增强、己糖通路及戊二醇通路的激活等[11]。自噬功能异常也在DN进程中发挥重要作用。DN的病理过程十分复杂,其不仅是由肾组织内单一细胞的病变而引发的,而是肾组织中的各个细胞都可能存在线粒体自噬障碍。本文将分别叙述肾足细胞、肾小球内皮细胞、系膜细胞以及肾小管上皮细胞中的线粒体自噬紊乱。
1 糖尿病肾病中肾足细胞的线粒体自噬紊乱
足细胞又称为肾小球脏层细胞,它是构成肾小球滤过系统的最后一道屏障,其损伤在糖尿病肾病的发展进程中起重要作用[12]。足细胞富含线粒体,在很大程度上依靠氧化磷酸化来供能,所以足细胞中线粒体功能紊乱也在DN进程中起重要的作用。很多研究表明,组蛋白去乙酰酶6(histone deacetylase Sirtuin 6,Sirt6)在一些疾病中能影响线粒体的形态和功能,使线粒体发生肿胀、空泡化和线粒体嵴消失,并且线粒体膜电位降低以及凋亡引起数量减少[13-14]。Sirt6在DN病理过程中起重要作用,能通过激活腺苷酸激酶(AMP kinase,AMPK)调控线粒体的生物合成和降解从而对线粒体起到保护作用。当Sirt6过表达时,高糖处理过的足细胞的p-AMPK表达增加,足细胞线粒体形态异常显著改善,活性氧(reactive oxygen species,ROS)产生降低,足细胞受损得到改善。鉴于Sirt6和AMPK在调控自噬活性中起重要作用,高糖环境下Sirt6和AMPK受抑制则会引起线粒体形态异常并最终引起线粒体自噬受抑制,从而在DN发病机制中起重要作用[15]。
高糖环境下及DN模型中,足细胞的PINK1,Parkin和LC3表达显著下降,而P62的表达显著升高,表明足细胞的线粒体自噬受到抑制[16-18]。研究发现,在足细胞核中叉头盒O1(forkhead-box class O1,FoxO1)能与PINK1基因的启动子结合,从而激活PINK1基因的转录,促进足细胞线粒体自噬。在高糖环境下,FoxO1被磷酸化而转移到胞浆中,丧失其转录活性,线粒体自噬从而受到抑制,线粒体ROS生成增加,线粒体膜电位降低,ATP水平降低。当足细胞中过表达FoxO1时,FoxO1能与PINK1基因的启动子相结合并激活PINK1的转录,使PINK1表达增加,从而激活PINK1和Parkin介导的线粒体自噬。一旦线粒体自噬被激活,则可恢复线粒体功能并改善足细胞内的氧化应激状态,从而改善DN的病理状态[19]。
2 糖尿病肾病中肾小球内皮细胞的线粒体自噬紊乱
高糖环境下,肾小球内皮细胞发生过度氧化应激、细胞内钙离子含量过高以及细胞骨架破坏等,从而导致肾小球内皮细胞选择透过性降低,引发蛋白尿的发生[20-21]。肾小球内皮细胞是肾小球滤过膜系统的内层,其损坏会引发炎症和蛋白尿的发生,故在DN的发生进程中起重要作用[22]。研究表明,在DN小鼠模型和高糖损伤肾小球内皮细胞模型中,线粒体形态和功能发生异常,ROS生成增加、线粒体膜电位降低。此外,PINK1和Parkin的表达降低,线粒体自噬受抑制并且细胞凋亡增加。而辅酶Q10(coenzyme Q10,CoQ10)能在一定程度上升高肾小球内皮细胞PINK1和Parkin的表达,激活线粒体自噬,从而在一定程度上改善DN。当采用线粒体自噬抑制剂Mdiv或siPINK1作用时,CoQ10的改善作用消失;并且当采用核转录因子E2相关因子2(nuclear factor E2-related factor 2,Nrf2)抑制剂ML385作用时,CoQ10的改善作用同样消失。表明CoQ10是通过Nrf2/抗氧化反应元件通路激活线粒体自噬,减少ROS生成,缓解线粒体形态异常,从而改善DN[23]。
3 糖尿病肾病中肾小球系膜细胞的线粒体自噬紊乱
肾小球系膜细胞是肾小球固有的细胞,肾小球系膜由系膜细胞和细胞外基质构成,细胞外基质包围细胞,细胞外基质数量增多以及发生肥大在DN进程中起重要作用。高糖可诱导肾小球系膜细胞中细胞外基质合成[24],使肾小球系膜细胞缺血受损而发展成肾小球硬化[25],肾小球硬化在DN进程中起重要作用。在以临床样本为对象的研究中发现,DN患者肾小球系膜细胞的PINK1和Parkin的mRNA含量相对正常肾小球系膜细胞显著降低,线粒体自噬水平也下降[26]。
在DN模型小鼠和高糖损伤肾小球系膜细胞模型中,肾小球系膜细胞线粒体过度分裂碎片化,ROS过度产生,线粒体通透性转换孔道开放从而使得线粒体依赖性细胞凋亡被激活[27]。同时,Parkin和LC3Ⅱ/LC3Ⅰ的表达降低,而P62的表达增加,线粒体自噬受抑制。研究表明,上述变化是由核受体NR4A1(nuclear receptor subfamily 4 group A member 1,NR4A1)通过激活线粒体分裂和抑制PINK1/Parkin介导的线粒体自噬引起的。在高糖环境下,系膜细胞中NR4A1表达增加从而激活P53的表达,而P53则会上调线粒体分裂因子的表达,从而激活线粒体分裂并下调Parkin的表达,进而抑制线粒体自噬。当敲除NR4A1基因时,线粒体分裂下降,氧化应激水平得以改善,线粒体通透性转换孔道开放受抑制,促凋亡蛋白进入胞浆减少,从而凋亡受到抑制。此外,PINK1/Parkin介导的线粒体自噬被激活使受损线粒体得以清除,细胞稳态得以维持,DN得以改善[28]。
在DN患者中晚期糖基化终末产物(advancedglycation endproducts,AGE)增多且清除降低。研究表明,AGE和高糖能促进肾小球系膜细胞线粒体膜电位去极化,使ROS产生增多,并且膜电位降低激活胱天蛋白酶9,而胱天蛋白酶9反过来激活胱天蛋白酶3从而引发系膜细胞凋亡[29]。ROS的过度产生及线粒体膜电位的去极化也会激活自噬及线粒体自噬,从而清除受损的线粒体,降低ROS水平,减少肾小球系膜细胞凋亡。而线粒体自噬受阻碍会引起系膜细胞凋亡增加,从而引起系膜细胞丢失,导致肾小球硬化[25,30]。
4 糖尿病肾病中肾小管上皮细胞的线粒体自噬紊乱
肾小管上皮细胞损伤会引起肾间充质炎症和纤维化,从而在DN进程中起重要作用。OPTN是线粒体自噬中重要的线粒体自噬受体,它包含泛素结合序列,能将具有多聚泛素链的线粒体和LIR结合,从而形成自噬体。核苷酸结合寡聚化结构域样受体3(nucleotide-binding oligomerization domainlike receptor family pyrin domain-containing 3,NLRP3)炎症小体能促进肾间充质炎症,从而在DN发展中起重要作用[31]。在DN中,线粒体碎片化产生过多的ROS,激活NF-κB信号通路,从而使NLRP3,白介素 1β前体(pro-interleukin-1 beta,pro-IL1β)和白细胞介素18(interleukin-18,IL-18)转录增加,NLRP3炎症小体被激活[32-33]。NLRP3炎症小体的激活在DN的发病机制中起重要的作用。研究表明,在高糖存在下,肾小管上皮细胞中动态相关蛋白1(dynamin-related protein-1,Drp-1)表达增加而线粒体融合蛋白2(mitofusin2,Mfn2)的表达降低,线粒体碎片化加剧,且线粒体膜电位降低。此外,高糖环境下,肾小管上皮细胞的NLRP3炎症小体表达增加,PINK1,Parkin和OPTN的表达下降,P62表达增加,线粒体自噬受到抑制。当过表达OPTN时,NF-κB信号通路受到抑制,且NLRP3炎症小体的激活受到抑制,线粒体自噬显著增加,线粒体ROS显著减少,受损的线粒体得以清除,DN从而得到改善[34-35]。
损坏的线粒体会产生过量ROS,造成线粒体氧化应激,并引起促凋亡因子释放而造成细胞凋亡,同样过量ROS和高糖环境也会抑制线粒体自噬途径,使细胞不能清除受损的线粒体,从而产生更多的ROS[36]。MitoQ是靶向线粒体的抗氧化剂,研究发现,其能增强高糖引起的肾小管上皮细胞的线粒体自噬缺陷,从而发挥对肾小管上皮细胞的保护作用,并且这种作用是通过Nrf/PINK1途径调控的[37-38]。在高糖环境下,肾小管上皮细胞中的Drp-1表达显著升高,而Mfn2的表达显著降低,此外线粒体的膜电位和ATP的活性也都降低,这表明线粒体受损,线粒体过度分裂,并且线粒体融合受到抑制。同时肾小管上皮细胞中LC3Ⅱ,PINK1,Parkin和Nrf2的表达显著下降,P62和Kelch样环氧氯丙烷相关蛋白1(Kelch-like ECH-associated protein,Keap1)表达显著升高,而细胞内和线粒体中的ROS显著增加,表明线粒体自噬受到抑制。当MitoQ处理高糖环境中的肾小管上皮细胞时,PINK1和Parkin表达升高,线粒体自噬水平升高,细胞损伤得到改善。而当沉默Keap1时,线粒体自噬水平升高作用增强;当沉默Nrf2时,线粒体自噬水平升高作用减弱,表明MitoQ是通过Nrf/PINK1途径调控PIN1/Parkin介导的线粒体自噬,从而增强线粒体自噬水平,改善肾小管上皮细胞损伤[39]。
5 结语
本综述从线粒体自噬在DN发病机制中的重要作用出发,介绍了哺乳动物中主要由PINK1和Parkin介导的线粒体自噬的具体机制。此外,还介绍了DN中肾组织各细胞中所发生的线粒体自噬紊乱,造成细胞损伤甚至死亡,最终促进DN的发生发展(图1)。虽已确定线粒体自噬在DN发病机制中起重要作用,但其在肾组织各细胞中的具体机制仍不明确,有待进一步探究。
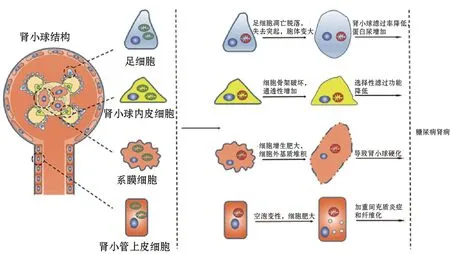
图1 肾组织中足细胞、肾小球内皮细胞、系膜细胞和肾小管上皮细胞4种细胞线粒体自噬紊乱.
DN发病早期难以诊断。目前,诊断DN的标志物是蛋白尿,但由于有些DN患者并未出现蛋白尿,使DN的诊断变得更加困难。目前,临床上用于治疗DN的药物主要包括降低心血管风险、控制血糖、控制血压及抑制肾素-血管紧张素系统的药物,但它们只能延缓DN的进展,并不能逆转DN所引起的病理变化。所以,现在迫切需要发现治疗DN的新靶点,旨在根治DN,降低DN的高发生率和死亡率。
线粒体为细胞提供生命所必需的能量,线粒体自噬紊乱会引起损伤的线粒体清除障碍,造成线粒体的形态异常和功能紊乱,从而引发机体疾病。增强线粒体自噬可恢复线粒体正常的形态和功能,并改善肾细胞的功能形态。所以,将线粒体自噬作为治疗DN的新靶点可能为治疗DN带来新的曙光。但当前由于线粒体自噬中的具体信号通路及作用机制还未阐明,所以目前通过改善线粒体自噬来治疗DN还有待继续研究。
猜你喜欢
安家(校外教育)(2022年6期)2022-01-03 11:47:06
世界科学技术-中医药现代化(2021年7期)2021-11-04 08:10:42
天津医科大学学报(2021年4期)2021-08-21 02:14:34
中成药(2018年6期)2018-07-11 03:01:04
中成药(2017年8期)2017-11-22 03:18:21
安徽医科大学学报(2016年12期)2017-01-15 14:21:58
医学研究杂志(2015年9期)2015-07-01 17:28:00
长江蔬菜(2015年3期)2015-03-11 15:10:29
中国药理学通报(2014年2期)2014-05-09 08:22:33
中国药理学通报(2014年2期)2014-05-09 08:22:26
