
心肌纤维化的表观遗传调控研究进展
2019-04-04 02:50:56占贞贞曾麒燕
中国比较医学杂志 2019年3期
汪 波,占贞贞,曾麒燕*
(1. 广西医科大学生物化学与分子生物学教研室,南宁 530021;2. 同济大学附属东方医院心力衰竭研究所,上海 200120)
心肌纤维化的典型特点是心肌组织中细胞外基质过度沉积,形成瘢痕组织,致使心肌失去正常的舒缩功能,以及影响心肌组织的电生理作用。心肌组织中的多种细胞在心肌纤维化的过程中发挥作用,包括心肌细胞、成纤维细胞、内皮细胞以及炎症细胞等[1]。成纤维细胞是心肌组织中含量最丰富的非心肌细胞,在心肌纤维化的过程中,其发挥的作用最为重要。当心脏暴露在病理条件下,如压力负荷、心肌梗死等,心肌成纤维细胞为维持心脏的正常结构而激活增殖,并转化为肌成纤维细胞,分泌大量的细胞外基质纤维化蛋白在心肌组织中过度沉积,最终致使心力衰竭的发生[2]。
目前研究发现多种机制可调节心肌纤维化,包括调节起始的炎症反应以及纤维化相关信号通路的激活从而达到控制纤维化反应的强度。在心肌压力应激反应及损伤中,常伴随着大量的心肌细胞坏死,坏死的心肌细胞释放大量的内源性危险因子,募集炎症细胞到达心肌组织。炎症细胞既可以清除坏死的心肌细胞及细胞外基质,也会分泌TGF-β等促纤维化因子[3]。TGF-β主要依赖于经典的TGF-β/smad通路,上调众多纤维化相关基因的表达,从而促进心肌的纤维化。多种信号调节分子参与TGF-β/smad通路的调控从而控制纤维化相关基因的表达,而近年来发现,表观遗传调控机制在心肌纤维化相关基因的表达调控中亦发挥着重要的作用。
表观遗传调控是指在不改变DNA碱基序列的前提下对基因的表达进行调节,其机制包括各种DNA修饰、组蛋白修饰、非编码RNA的调控作用等。核小体是染色质的基本结构,由147 bp的DNA缠绕在组蛋白上形成,其中组蛋白又包括H2A、H2B、H3和H4等形成的八聚体,通过对组蛋白进行修饰,改变染色质构象,从而达到对基因的表达实现表观遗传调控。近年来发现DNA甲基化修饰、组蛋白修饰以及microRNA等表观遗传机制参与了心肌纤维化的发生发展过程。
1 DNA甲基化与心肌纤维化
DNA甲基化是调节基因表达的重要表观遗传修饰途径。DNA甲基化主要集中在CpG二核苷酸的胞嘧啶5号碳原子上,散在存在于哺乳动物基因组中约70% CpG胞嘧啶存在甲基化修饰[4],另外存在一些片段长度在200 bp以上,成簇存在于哺乳动物启动子中或靠近启动子区域的CpG被称之为CpG岛,与散在存在的CpG相反,CpG岛通常为非甲基化状态,但在机体处于异常或疾病状态时,CpG岛会发生甲基化,从而使相关基因沉默。DNA甲基化依赖于DNA甲基化转移酶(DNA methyltransferases,DNMTs)的协助,哺乳动物主要表达三类DNMTs,分别为DNMT3A、DNMT3B及DNMT1A,其中DNMT1A主要涉及维持DNA甲基化模式,DNMT3A/B则起到从头催化DNA甲基化的作用[5]。
研究发现,心肌纤维化的发生与多种基因的DNA甲基化有关(图1)。缺氧可通过缺氧诱导因子1α(hypoxia inducible factor 1 subunit alpha,HIF-1α)诱导DNA超甲基化,并且促进DNA甲基化转移酶DNMT1、DNMT3B表达,通过siRNA敲低DNMT3B的表达后能显著降低胶原蛋白1和α平滑肌激动蛋白的表达;此外,利用DNMT的抑制剂也可以抑制TGF-β促纤维化的作用[6]。Tao等[7]的研究发现,在心肌成纤维细胞激活的过程中RASSF1A的表达降低,并且伴随着DNMT3A的过表达,而DNMT3A可抑制RASSF1A的表达,并最终导致纤维化的发生。另外,过度的自噬可加剧心肌肥大以及心肌纤维化,研究显示DNMT3A可直接抑制miR-200b的表达,促进小鼠主动脉缩窄术模型小鼠心脏自噬的发生,并最终影响着心肌纤维化的发展[8]。miR-369-5p过表达可抑制心肌成纤维细胞的增殖以及心肌纤维化水平,这一过程是通过miR-369-5p直接抑制DNMT3A的表达而使Patched1超甲基化抑制失活实现的[9]。RASAL1是一种能抑制Ras信号通路活化的Ras-GTP酶,研究显示在纤维化心脏中RASAL1的启动子区域超甲基化,使Ras信号通路激活增加引起心肌纤维化,而通过TET依赖的RASAL1启动子去甲基化作用可逆转TGF-β诱导的纤维化[10]。由此可见,DNA甲基化相关的修饰酶通过直接或间接的机制途径,影响纤维化相关基因的表达,从而调控心肌纤维化的程度,但是所调控的靶基因是否存在特异性,以及是否还有其他的核蛋白共同参与调控过程,需要进一步深入研究。
2 组蛋白修饰与心肌纤维化
组蛋白是染色质的重要组成,组蛋白尾的氨基酸末端从核小体突出,为相关修饰提供多种表观遗传学修饰位点,从而调控基因的转录表达。组蛋白修饰方式主要包括乙酰化、甲基化、泛素化、磷酸化以及SUMO化等[11]。与DNA修饰相似,组蛋白修饰也由不同的修饰酶介导,包括去乙酰/甲基化酶、乙酰/甲基转移酶等。在心肌纤维化过程中,研究发现组蛋白乙酰化、甲基化和磷酸化修饰及其对应的修饰酶与心肌纤维化的发生发展密切相关(图1)。
2.1 组蛋白乙酰化修饰
组蛋白乙酰化的状态取决于严格控制其平衡的两种组蛋白修饰酶:组蛋白乙酰转移酶(histone acetyltransferases,HATs)和组蛋白去乙酰化酶(histone deacetylases,HDACs)。HATs催化向核小体组蛋白上的赖氨酸残基加入乙酰基团,导致染色质松弛,使DNA与转录因子及其他调控因子的亲和力更强,促进DNA的转录。而HDACs催化乙酰基团从赖氨酸处分离,导致染色质更为紧密,从而抑制基因的转录[12]。
根据在细胞中的定位,HATs可分为两类:A型位于核内,是核小体组蛋白乙酰化的主要催化酶类,B型定位于细胞质,主要作用是乙酰化新合成的非组蛋白如某些信号分子蛋白[13]。关于HATs在心肌纤维化中的研究仍然有限。目前已有研究表明P300与心肌纤维化有关,如CTRP3(C1q and TNF related 3)可通过抑制P300与SMAD3之间的作用从而降低心肌纤维化[14]。组蛋白乙酰转移酶KAT8(lysine acetyltransferase 8,又称为MOF)可以松弛NOX(NADPH oxidase)启动子周围的染色质结构从而激活NOX的转录,在敲除MOF后可显著降低缺氧/复氧诱导的NOX转录激活、ROS的释放、心肌梗死及后期的纤维化面积[15],因此MOF是缺血再灌注损伤诱导的心肌纤维化中重要的调节分子。然而,其他的组蛋白乙酰转移酶是否通过靶向组蛋白或非组蛋白从而参与心肌纤维化尚待探索。
对于HDACs,目前在哺乳动物体内共发现四类18种,分别为Ⅰ型去乙酰化酶(HDAC 1 ~ 3,8)、Ⅱ型去乙酰化酶(HDAC 4 ~ 7,9,10)、Ⅲ型去乙酰化酶(SIRT,SIRT 1 ~ 7)和只在人类中发现的第Ⅳ型去乙酰化酶HDAC11。研究发现,选择性抑制Ⅰ型HDACs后可阻断心肌成纤维细胞的细胞周期进程,解除周期蛋白依赖性激酶抑制因子P15和P57基因的抑制,最终可有效抑制血管紧张素Ⅱ诱导的心肌纤维化[16]。另据报道,在充血性心衰中HDAC1和HDAC2表达升高,通过小分子抑制剂Mocetinostat抑制其活性后,可显著降低小鼠充血性心衰模型心脏中CD90阳性心肌肌成纤维细胞的激活而逆转心肌纤维化[17]。SAHA是一种广谱HDAC抑制剂,SAHA处理DOCA-salt高血压模型大鼠可有效抑制高血压诱导的心肌纤维化[18]。ITF2357是一种针对多种疾病并进入到三期临床实验的HDAC抑制剂,在Dahl盐敏感大鼠模型中,ITF2357可显著抑制与心室舒张功能障碍有关的心肌结构变化,包括心肌肥厚以及心肌纤维化水平[19]。SIRT2的蛋白表达在心肌肥厚心脏组织中显著降低,研究发现Sirt2基因敲除小鼠中血管紧张素诱导的心肌纤维化显著加重,而心脏特异性过表达SIRT2可通过直接激活AMPK下游的激酶LKB1从而抑制血管紧张素诱导的心肌纤维化[20]。另有研究表明HDAC6、SIRT6(sirtuin 6)和SIRT3(sirtuin 3)等都可通过不同的机制参与心肌纤维化进程的调控[21 - 23]。因此,针对组蛋白去乙酰化酶开发更为有效和特异的抑制剂对于心肌纤维化相关疾病的临床治疗研究具有重要的价值。
2.2 组蛋白甲基化修饰
组蛋白上的赖氨酸和精氨酸残基的甲基化修饰更为复杂,赖氨酸可以发生单甲基化、双甲基化和三甲基化修饰,精氨酸可以被单甲基化、双甲基化[24]。根据组蛋白甲基化位点的不同,可导致不同的基因表达状态,如H3K4me3、H3K36me3和H3K79me3等可促使基因的转录的激活,而H3K9me3、H3K27me3及H4K20me3等可导致基因转录的抑制[25]。另外,组蛋白同一位点甲基化的不同修饰状态也可导致基因转录水平的不同,例如H3K9me可促进基因的转录,而H3K9me3则抑制基因的表达[26]。
组蛋白残基甲基化水平的动态平衡是由甲基转移酶和去甲基化酶维持的。组蛋白赖氨酸甲基转移酶至少可分为8个家族共33个成员,包括KMT1-KMT8,其中只有KMT4(lysine methyltransferase 4)缺少典型的SET催化结构域[27]。G9a是一种能够催化H3K9单/双甲基化的甲基转移酶,研究发现G9a可与MEF2C(myocyte enhancer factor 2C)形成复合体并结合异染色质抑制抗心肌肥厚基因的表达,G9a敲除后心肌纤维化水平显著升高,促进心肌肥厚[28]。血管紧张素Ⅱ可诱导内皮细胞SET1(SET domain containing 1)的表达,SET1被募集到内皮素1启动子区域促进内皮素1的转录,导致心肌肥厚及纤维化水平增加,通过内皮细胞特异敲除SET1可显著降低血管紧张素Ⅱ诱导的心肌肥大及心肌纤维化[29]。
组蛋白去甲基化酶已发现有20多种,根据同源性可分为7个亚家族,包括KDM1(lysine demethylase 1)、KDM2/7、KDM3、KDM4、KDM5和KDM6[30]。以往的研究发现KDM4A(lysine demethylase 4 A)缺失可显著改善主动脉缩窄诱导的小鼠心肌纤维化程度及心肌肥大[31]。PHF8(PHD finger protein 8)可催化H3K9me1/2和H4K20me1组蛋白去甲基化,Liu等[32]的研究发现PHF8可通过抑制Akt-mTOR通路从而抑制主动脉缩窄诱导的心肌肥厚,并且在PHF8转基因小鼠中TAC诱导的纤维化水平显著降低。但是,对于其他种类的组蛋白甲基化或去甲基化酶,其在心肌纤维化中的功能有待进一步研究,而且也有待开发相应的抑制剂或激动剂,以用于心肌纤维化相关疾病的临床治疗研究。
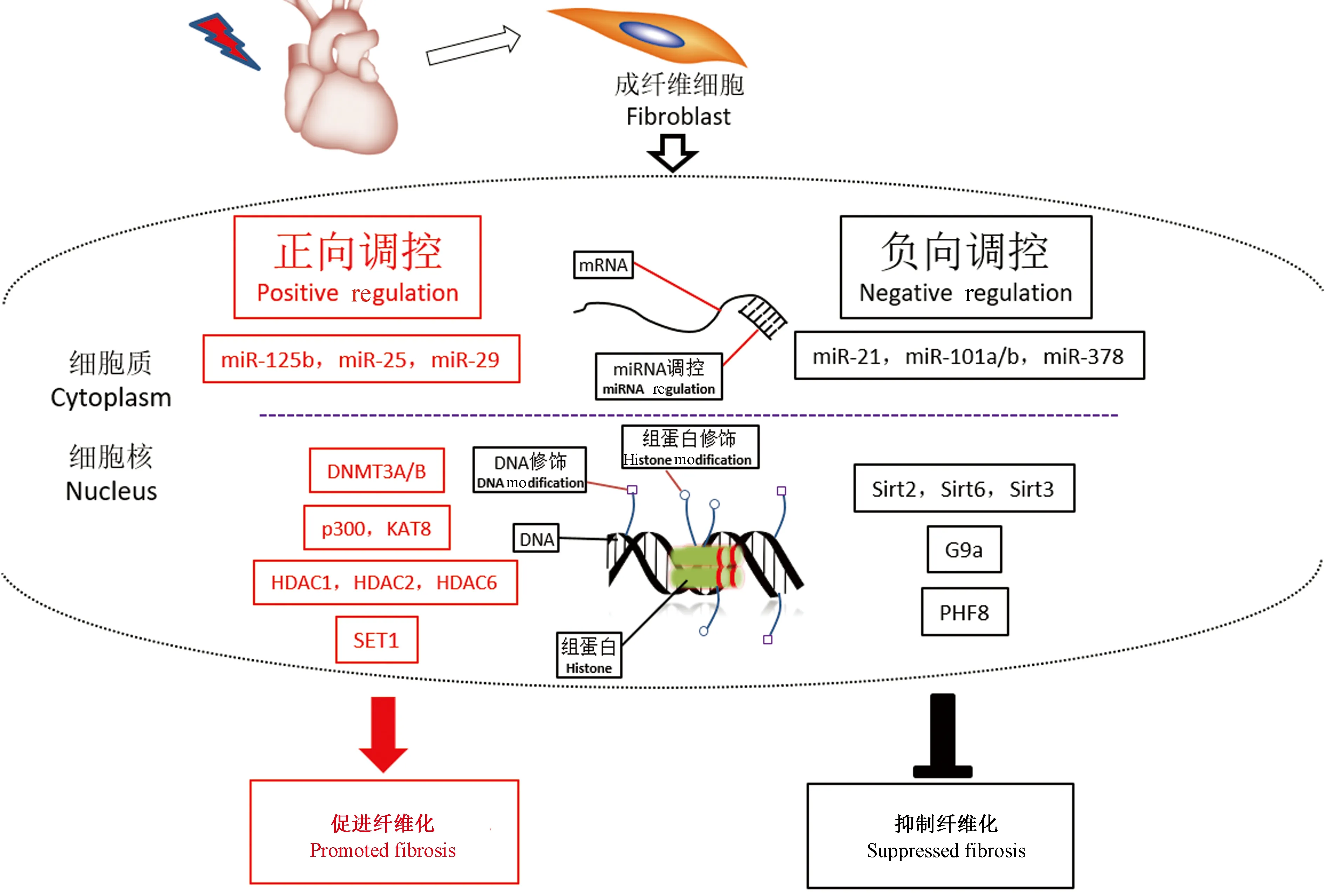
图1 心肌纤维化的表观遗传学调控Figure 1 Epigenetic regulation of myocardial fibrosis
3 MicroRNA与心肌纤维化
MicroRNA是一类具有19 ~ 25个核苷酸序列的短链非编码RNA。MicroRNA可与目的mRNA的3’ UTR区互补结合,从而降解mRNA或抑制mRNA的翻译,最终影响蛋白质的合成[33]。MicroRNA分子在不同物种中高度保守,是多种细胞生理病理过程包括发育、增殖、分化、凋亡和应激反应等的重要调节因子[34]。越来越多的研究表明特定microRNA的表达与心肌纤维化的发生发展有着密不可分的关系,在心脏疾病中,microRNA作为潜在的诊断标志及治疗靶点越来越受到人们的关注(图1)。
在人及小鼠纤维化心脏中miR-125b表达显著升高,研究发现miR-125b可抑制p53介导的成纤维细胞增殖,体内抑制miR-125b的表达可降低纤维化相关基因的表达,并最终降低血管紧张素Ⅱ诱导的心肌纤维化水平[35]。在主动脉缩窄小鼠模型中,抑制miR-21的表达可降低激酶ERK的活性,从而抑制心肌间质纤维化以及心功能障碍[36]。另一项研究发现,通过纳米材料靶向心梗后心脏巨噬细胞过表达miR-21,使其转变为抗炎性巨噬细胞,最终可降低心梗后纤维化水平[37]。其他更多的研究表明miR-25和miR-29等促进压力负荷以及心肌梗死诱导的心肌纤维化,而miR-101a/b和miR-378等可通过不同的作用途径抑制心脏纤维化的进程[38 - 42]。这些研究表明miRNA的确参与心肌纤维化的病理过程调控,但是是否能成为心肌纤维化相关疾病的诊断标志物或者治疗的靶标,还需深入探索。
4 展望
尽管心脏纤维化过程中表观遗传调控的研究取得了一定的进展,但还存在着许多未知之处。近年来随着高通量甲基化测序等新技术体系的出现和完善,为研究表观遗传调控在心肌纤维化病理生理学中的作用提供新的有效手段。目前越来越多的新的表观修饰方式被发现,如在组蛋白氨基酸残基在上还有巴豆酰化、丙酰化、丁酰化、琥珀酰化等全新的修饰类型,在RNA上存在m6A、m1A及m5C等新型修饰,以及染色质三维空间结构的改变等,这些新型表观修饰方式的阐明将大大的拓展我们在心脏疾病中的研究思路。最近的研究就发现RNA的m6A去甲基化酶FTO(alpha-ketoglutarate dependent dioxygenase)参与心肌纤维的调控[43],那么其他的新发现的表观修饰方式是否也在心肌纤维化中发挥调节作用将是新的研究方向。另外,这些众多的表观遗传修饰机制是协同还是竞争性的参与调节不同心脏疾病诱导的心肌纤维化过程,又是哪些因素启动这些表观遗传修饰,这些问题都有待解决。这些表观机制的阐明将有助于我们针对各种介导表观修饰的表观酶分子,开发相应的抑制剂或激动剂,从而用于具有巨大的潜力和前景的心肌纤维化相关疾病的临床治疗应用研究。
猜你喜欢
中国生物化学与分子生物学报(2022年8期)2022-09-08 00:39:50
河北果树(2021年4期)2021-12-02 01:14:50
昆明医科大学学报(2021年8期)2021-08-13 08:59:50
云南医药(2021年3期)2021-07-21 05:40:30
上海公路(2019年3期)2019-11-25 07:39:28
福建基础教育研究(2019年10期)2019-05-28 08:27:04
中国组织化学与细胞化学杂志(2016年4期)2016-02-27 11:15:53
中国现代医学杂志(2015年26期)2015-12-23 11:04:22
中国当代医药(2015年33期)2015-03-01 02:09:20
中国当代医药(2015年16期)2015-03-01 02:03:13
