
一测多评法同时测定参芪二仙片中9种成分
2019-04-01 12:43蔡楚兰
中成药 2019年3期
蔡楚兰, 廖 勇
(1.宜昌市优抚医院科教科,湖北宜昌443005;2.三峡大学第二临床医学院,三峡大学附属仁和医院,湖北 宜昌443001)
参芪二仙片收载于中华人民共和国卫生部药品标准 (中药成方制剂第二十册)[1],由淫羊藿、酒制仙茅、盐制巴戟天、红参、黄芪、当归、盐制黄柏、盐制知母8味药材组成,具有补肾填精、调补冲任、益气养血的功效,临床上主要用于治疗肾虚腰膝酸软、阳痿早泄、遗精、更年期经血不调等症,但其现行质量标准未对方中成分进行定量测定, 仅赵玉华[2]、 杨国振[3]等采用 HPLC 法测定淫羊藿苷含有量,难以有效控制产品质量稳定性和临床疗效一致性。因此,本实验采用一测多评法,以参芪二仙片中淫羊藿苷为内标,计算其与仙茅苷、仙茅苷乙、水晶兰苷、去乙酰基车叶草苷酸、朝藿定A、朝藿定B、朝藿定C、宝藿苷Ⅰ的相对校正因子,测定各成分含有量,以期为该制剂质量控制提供参考。
1 材料
Agilent 1100型高效液相色谱仪 (美国安捷伦公司);BP211D型电子分析天平 (瑞士Mettler-Toledo公司);KQ2200型超声波清洗器 (昆山市超声仪器有限公司)。参芪二仙片 (批号161002、161103、170107),购于沈阳红药集团股份有限公司。仙茅苷 (110771-201707)、水晶兰苷 (111870-201303)、朝藿定 C (111780-201503)、淫羊藿苷 (110737-201516)、宝藿苷Ⅰ (111852-201603)对照品购于中国食品药品检定研究院;仙茅苷乙 (143601-09-6)对照品购于深圳市佰森生物技术有限公司;去乙酰基车叶草苷酸 (14259-55-3)、朝藿定 A (110623-72-8)、朝藿定B(110623-73-9)对照品购于上海纯优生物科技有限公司。乙腈为色谱纯;其他试剂均为分析纯。
2 方法与结果
2.1 色谱条件 Kromasil C18色谱柱 (4.6 mm×250 mm, 5 μm); 流动相乙腈 (A) -0.1% 冰醋酸(B), 梯度洗脱 (0~16 min, 10.0%A; 16~34 min,10.0%→21.0%A;34~47 min,21.0%→26.0%A;47~58 min,26.0% → 45.0%A;58~65 min,45.0%→10.0%A); 0~23 min 时在280 nm[4]波长下检测仙茅苷和仙茅苷乙,23~34 min时在235 nm[5-6]波长下检测水晶兰苷和去乙酰基车叶草苷酸,34~65 min 时在 270 nm[2-3,7-11]波长下检测朝藿定 A、 朝藿定B、朝藿定C、淫羊藿苷、宝藿苷Ⅰ;体积流量0.9 mL/min; 柱温30℃; 进样量10 μL。
2.2 溶液制备
2.2.1 供试品溶液 取片剂10片,除去糖衣后研细,精密称取细粉约0.4 g,置于具塞锥形瓶中,精密加入 70%乙醇 50 mL,称定质量,超声(100 W、40 kHz)30 min,放冷,70%乙醇补足减失的质量,摇匀,过滤,即得。
2.2.2 对照品溶液 精密称取仙茅苷、仙茅苷乙、水晶兰苷、去乙酰基车叶草苷酸、朝藿定A、朝藿定B、朝藿定C、淫羊藿苷、宝藿苷Ⅰ对照品适量,70%乙醇制成质量浓度分别为0.432、0.176、0.814、 0.498、 0.262、 0.556、 1.038、 0.642、0.416 mg/mL的贮备液,各精密吸取2.5 mL,置于50 mL量瓶中,70%乙醇稀释至刻度,制成质量浓度分别为21.6、8.8、40.7、24.9、 13.1、27.8、51.9、 32.1、 20.8 μg/mL 的溶液, 即得。
2.2.3 阴性样品溶液 按片剂质量标准项下的处方和工艺,分别制成不含仙茅、巴戟天、淫羊藿的阴性样品,按 “2.2.1”项下方法制备,即得。
2.3 系统适用性考察与专属性试验 精密吸取供试品、对照品、阴性样品溶液适量,在 “2.1”项色谱条件下进样测定,结果见图1。由图可知,各成分与其相邻色谱峰的分离度均大于1.5,理论塔板数按各色谱峰计均大于3 500,阴性无干扰。
2.4 线性关系考察 精密吸取 “2.2.2”项下对照品溶液各 2.0 mL,置于同一20 mL量瓶中,70%乙醇定容至刻度,摇匀,作为混标液A,精密吸取适量,70%乙醇分别稀释 2、4、8、16、20倍,作为混标液B、C、D、E、F,精密吸取适量,在 “2.1”项色谱条件下进样测定。以成分质量浓度为横坐标 (X),峰面积为纵坐标 (Y)进行回归,结果见表1,可知各成分在各自范围内线性关系良好。
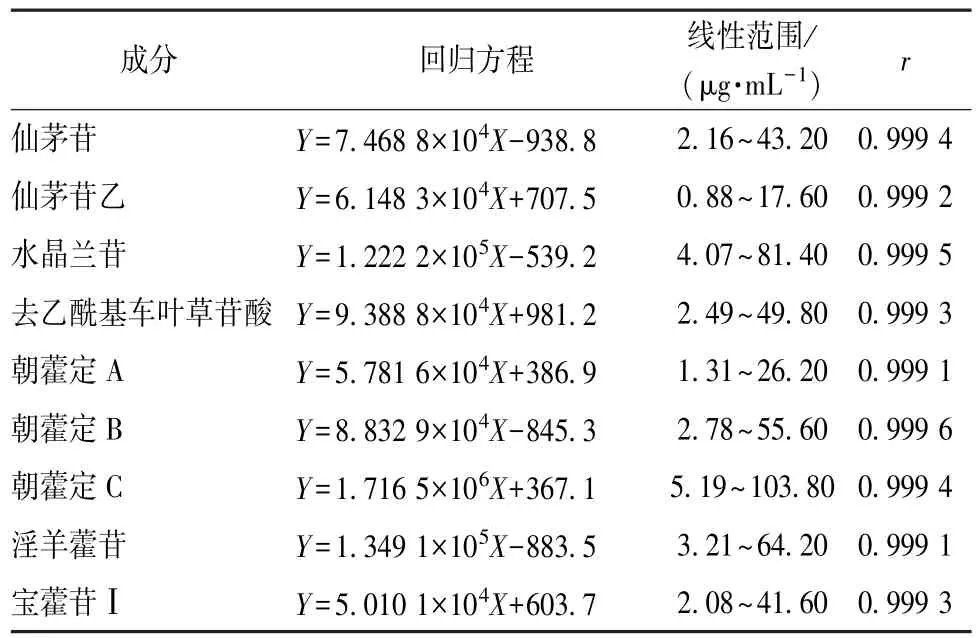
表1 各成分线性关系Tab.1 Linear relationships of various constituents
2.5 方法学考察

图1 各成分HPLC色谱图Fig.1 HPLC chromatograms of various constituents
2.5.1 精密度试验 取 “2.2.3”项下对照品溶液,在 “2.1”项色谱条件下进样测定6次,记录峰面积,考察日内精密度;连续进样6 d,每天1次,考察日间精密度。结果,仙茅苷、仙茅苷乙、水晶兰苷、去乙酰基车叶草苷酸、朝藿定A、朝藿定B、朝藿定C、淫羊藿苷、宝藿苷Ⅰ日内峰面积RSD分别为 0.91%、1.13%、0.66%、0.80%、1.09%、0.87%、0.59%、0.64%、0.93%,日间峰面积 RSD分别为 1.08%、1.27%、0.74%、0.85%、1.03%、0.99%、0.57%、0.72%、1.01%,表明该方法精密度良好。
2.5.2 重复性试验 取同一批片剂,按 “2.2.1”项下方法制备供试品溶液6份,在 “2.1”项色谱条件下进样测定,测得仙茅苷、仙茅苷乙、水晶兰苷、去乙酰基车叶草苷酸、朝藿定A、朝藿定B、朝藿定C、淫羊藿苷、宝藿苷Ⅰ含有量RSD分别为 0.32%、 1.54%、 1.16%、 1.23%、 0.49%、1.30%、1.01%、0.76%、1.36%,表明该方法重复性良好。
2.5.3 稳定性试验 取同一批片剂,按 “2.2.1”项下方法制备供试品溶液,室温下于0、2、4、6、8、12 h在 “2.1”项色谱条件下进样测定,测得仙茅苷、仙茅苷乙、水晶兰苷、去乙酰基车叶草苷酸、朝藿定A、朝藿定B、朝藿定C、淫羊藿苷、宝藿苷Ⅰ峰面积 RSD分别为 0.88%、1.15%、0.69%、 0.74%、 1.06%、 0.85%、 0.57%、0.62%、1.09%,表明溶液在12 h内稳定性良好。
2.5.4 加样回收率试验 取含有量已知的片剂适量,除去糖衣后研细,精密称取细粉6份,每份约0.2 g,置于 50 mL量瓶中,精密加入仙茅苷(0.257 mg/mL)、 仙茅苷乙 (0.098 mg/mL)、 水晶兰苷 (0.553 mg/mL)、去乙酰基车叶草苷酸(0.287 mg/mL)、 朝藿定 A (0.151 mg/mL)、 朝藿定 B (0.331 mg/mL)、 淫羊藿苷 (0.394 mg/mL)、宝藿苷Ⅰ (0.227 mg/mL) 对照品溶液各1.0 mL,以及朝藿定 C(0.353 mg/mL)对照品溶液2.0 mL,按 “2.2.3”项下方法制备供试品溶液,在 “2.1”项色谱条件下进样测定,计算回收率。结果,仙茅苷、仙茅苷乙、水晶兰苷、去乙酰基车叶草苷酸、朝藿定A、朝藿定B、朝藿定C、淫羊藿苷、宝藿苷Ⅰ平均加样回收率分别为98.63%、97.13%、99.41%、98.38%、97.46%、98.49%、100.05%、 99.19%、 97.70%, RSD 分别为1.18%、 1.44%、 0.86%、 1.25%、 1.52%、0.94%、0.85%、1.03%、1.32%。
2.6 相对校正因子计算 精密吸取 “2.4”项下混标液适量,在 “2.1”项色谱条件下进样测定,以淫羊藿苷为内标,计算其他8种成分相对校正因子 fk/s, 公式为 fk/s=fk/fs= (WkAs) /(WsAk) (Wk为内标质量浓度,Ak为内标峰面积,Ws为其他成分质量浓度,As为其他成分峰面积),结果见表2。

表2 各成分相对校正因子Tab.2 Relative correction factors of various constituents
2.7 各因素对相对校正因子的影响
2.7.1 仪器、色谱柱 取 “2.2.3”项下对照品溶液适量,在 “2.1”项色谱条件下进样测定,考察Agilent 1100、LC-20AT色谱仪和 Kromasil C18、Agilent TC-C18、Diamonsil C18色谱柱对相对校正因子的影响,结果见表3,可知均无明显影响。

表3 不同仪器、色谱柱对相对校正因子的影响Tab.3 Effects of different instruments and columns on relative correction factors
2.7.2 体积流量 取 “2.2.3”项下对照品溶液适量,在 “2.1”项色谱条件下进样测定,考察体积流量0.8、0.9、1.0 mL/min对相对校正因子的影响,结果见表4,可知均无明显影响。

表4 不同体积流量对相对校正因子的影响Tab.4 Effects of different volume flow rates on relative correction factors
2.7.3 柱温 取 “2.2.3”项下对照品溶液适量,在 “2.1”项色谱条件下进样测定,考察柱温25、30、35℃对相对校正因子的影响,结果见表5,可知均无明显影响。
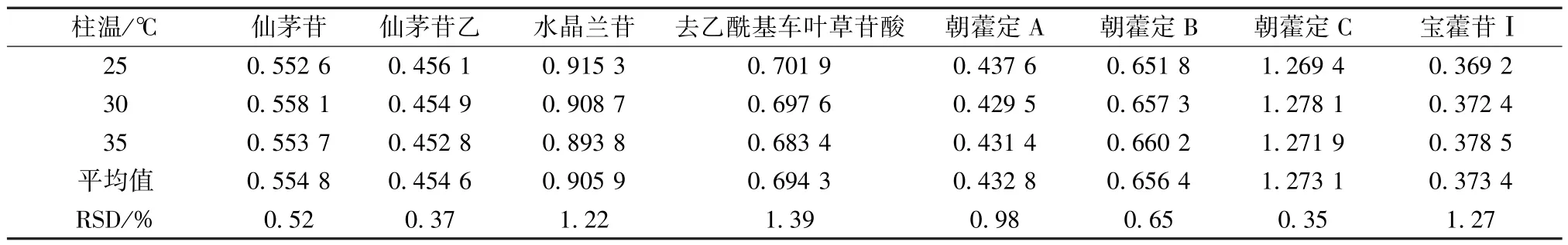
表5 不同柱温对相对校正因子的影响Tab.5 Effects of different column temperatures on relative correction factors
2.8 色谱峰定位 本实验采用相对保留值法对待测成分色谱峰进行定位,结果见表6,可知不同仪器、色谱柱下各成分相对保留值无明显差异。
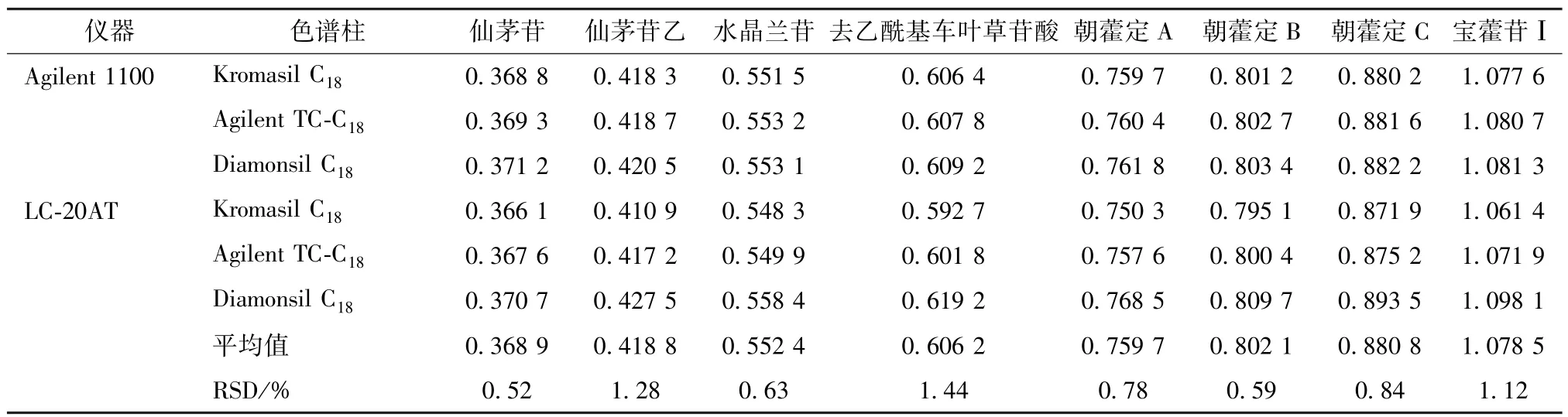
表6 各成分相对保留值Tab.6 Relative retention values of various constituents
2.9 样品含有量测定 取3批片剂 (批号161002、161103、 170107) 适量, 按 “2.2.1” 项下方法制备供试品溶液,在 “2.1”项色谱条件下进样测定,分别采用外标法和一测多评法计算含有量,结果见表7,可知2种方法所得结果无明显差异,相对平均偏差 (RAD)均<2%。
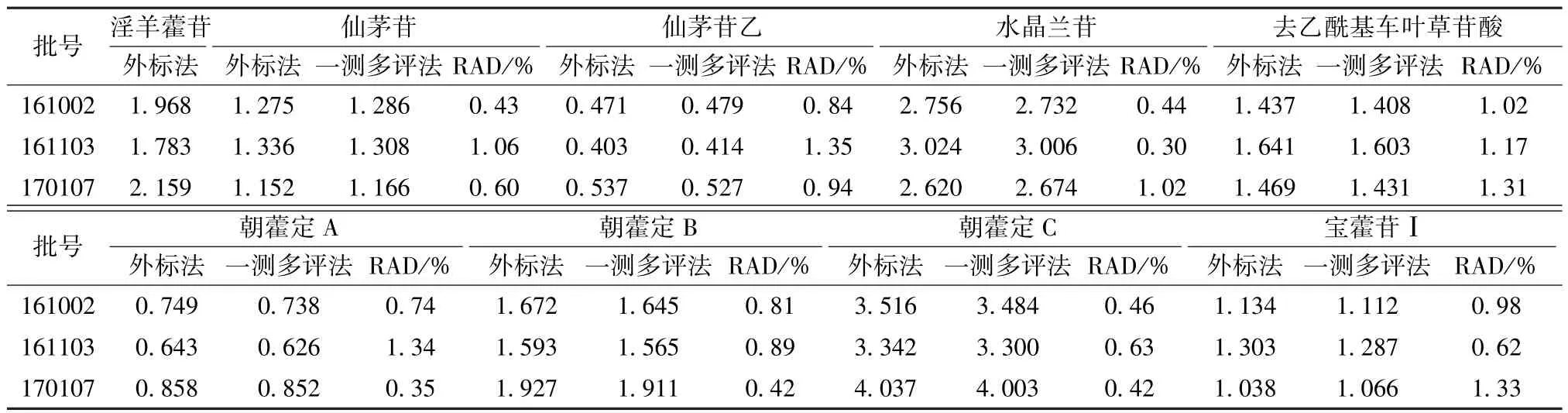
表7 各成分含有量测定结果 (mg/g)Tab.7 Results of content determination of various constituents (mg/g)
3 讨论
3.1 供试品溶液制备方法选择 本实验考察了提取溶剂 (50% 乙醇[2-3,7,9-12]、 70% 乙醇[5,13]、 乙醇、50% 甲醇、 甲醇[4,10])、 提取方式 (超声[2,5,7,9-10]、加热回流[3-4,8]) 对待测成分提取效果的影响,发现以70%乙醇为提取溶剂时综合提取效率最佳,而超声、加热回流提取所得结果差异不大,但前者更方便稳定。然后,又考察了提取时间 (15、30、60 min)对提取效率的影响,同时对超声功率、频率也进行了不断摸索。最终确定,供试品溶液制备方法为以70%乙醇为提取溶剂,超声 (100 W、40 kHz) 30 min。
3.2 流动相选择 本实验以待测成分峰形及分离效果、色谱图基线平稳情况、出峰时间为指标,考察了 乙腈-水[2-3,7,9-11,13]、 甲 醇-0.1% 磷 酸[5,10]、 乙腈-0.1%冰醋酸流动相体系[4,8], 并对流动相洗脱比例进行了不断摸索。最终确定,以乙腈-0.1%冰醋酸为流动相,在 “2.1”项色谱条件下梯度洗脱。
4 结论
中成药复方制剂具有多成分、多靶点、协同作用的特点,单一成分难以实现对其整体质量的评价和控制,故多指标质量控制已成为的必然趋势,但它又存在对照品使用量大、单体不稳定或价格昂贵、检验成本高等问题。一测多评法根据中药不同有效成分之间存在的内在函数关系和比例,通过测定某1种性质稳定、对照品价廉的成分来实现多种成分的同步测定,可大大降低检验成本,提高检测结果准确性。本实验采用该方法同时测定参芪二仙片中仙茅苷、仙茅苷乙、水晶兰苷、去乙酰基车叶草苷酸、朝藿定A、朝藿定B、朝藿定C、淫羊藿苷、宝藿苷Ⅰ的含有量,所建立的相对校正因子可信度较高,而且该方法简便准确,可为提高该制剂质量标准提供有力的数据支持。
猜你喜欢
色谱(2022年11期)2022-11-10
色谱(2022年10期)2022-10-13
天津中医药大学学报(2022年4期)2022-08-27
青岛大学学报(医学版)(2022年3期)2022-08-05
现代中药研究与实践(2022年1期)2022-03-19
食品安全导刊(2021年21期)2021-08-30
国学(2020年1期)2020-06-29
家庭影院技术(2018年11期)2019-01-21
中国骨质疏松杂志(2019年1期)2019-01-06
分析化学(2017年12期)2017-12-25
