
以巨噬细胞活化综合征起病的儿童系统性红斑狼疮1例报告并文献复习
2019-03-28 06:39姚坤宇刘聪聪李善玉
山西医科大学学报 2019年3期
姚坤宇,刘聪聪,张 丽,兰 坤,李善玉
(吉林大学第一医院儿科,长春 130000;*通讯作者,E-mail:shanyul@sina.com)
巨噬细胞活化综合征(macrophage activation syndrome,MAS)是一种继发性噬血细胞综合征(hemophagocytic syndrome,HLH),常见于全身型幼年特发性关节炎(systemic onset juvenile idiopathic arthritis,SOJIA),病死率高,且发病率逐年增高[1]。系统性红斑狼疮(systemic lupus erythematosus,SLE)是一种多系统受累的自身免疫性疾病,其临床表现除发热、皮疹等共同表现外,因受累器官不同而表现不同。Boone在1976年第一届ARA会议报告的1例SOJIA患儿因肝衰竭死亡的病例是儿科风湿系统疾病合并MAS的最早报道,1993年Stephan等[2]提出MAS的概念,受到临床医师的广泛关注。近年来国内外有报道MAS可继发于SLE,儿童SLE合并MAS早期表现常不典型且病情进展迅速、预后不良,所以尽早诊断及治疗十分重要,本例患者以MAS起病,入院时高度怀疑SLE,就诊后2周达到免疫指标。现将本例以MAS起病的儿童SLE诊治经过及文献复习报道如下。
1 病例资料
患儿,男,8岁,因间断发热半个月、发现血细胞减少2 d于2017年7月22日入住吉林大学第一医院小儿风湿科。间断发热,热型不规则,呈中等度热,伴左面部皮疹,无痒感,当地医院血常规三系减少、肝功谷丙转氨酶升高。患儿近期精神状态欠佳,无结膜充血、畏光,无杨梅舌,无关节肿痛。既往体健。入院查体:精神状态欠佳,颜面部可见红色皮疹,以两侧面颊部及左侧嘴角下皮肤为著(见图1),无贫血貌,颈部可触及数枚肿大的淋巴结,最大者约1.5 cm×1.2 cm,活动可,无压痛,口腔黏膜多处溃疡,最大约0.5 cm×0.5 cm。腹软,肝肋下约4 cm,脾肋下约1.0 cm,四肢及关节无肿痛,余查体未见明显异常。辅助检查:入院血常规白细胞1.96×109/L,红细胞3.79×1012/L[(4.3-5.8)×1012/L],血红蛋白103 g/L,血小板99×109/L。血沉27 mm/1 h(0-15 mm/1 h),铁蛋白3 148.2 μg/L(20-200 μg/L);乳酸脱氢酶988 U/L(135-266 U/L),肝功:门冬氨酸氨基转移酶100.3 U/L(0-40 U/L),丙氨酸氨基转移酶58.9 U/L(0-40 U/L)。三酰甘油正常;ANA系列、补体两项、凝血常规正常;EB病毒抗体及核酸定量正常,血清二价铁5.4 μmol/L(9-22 μmol/L),总铁结合力正常,类风湿因子、抗链球菌溶血素O、Coomb’s试验、尿便常规、心肌酶、肾功、外科综合、出血热抗体、心脏彩超正常。肺部CT平扫:①考虑左肺下叶炎性小结节;②颈根部肌间隙、纵隔及双侧腋窝多发淋巴结。腹部彩超:肝、脾增大。
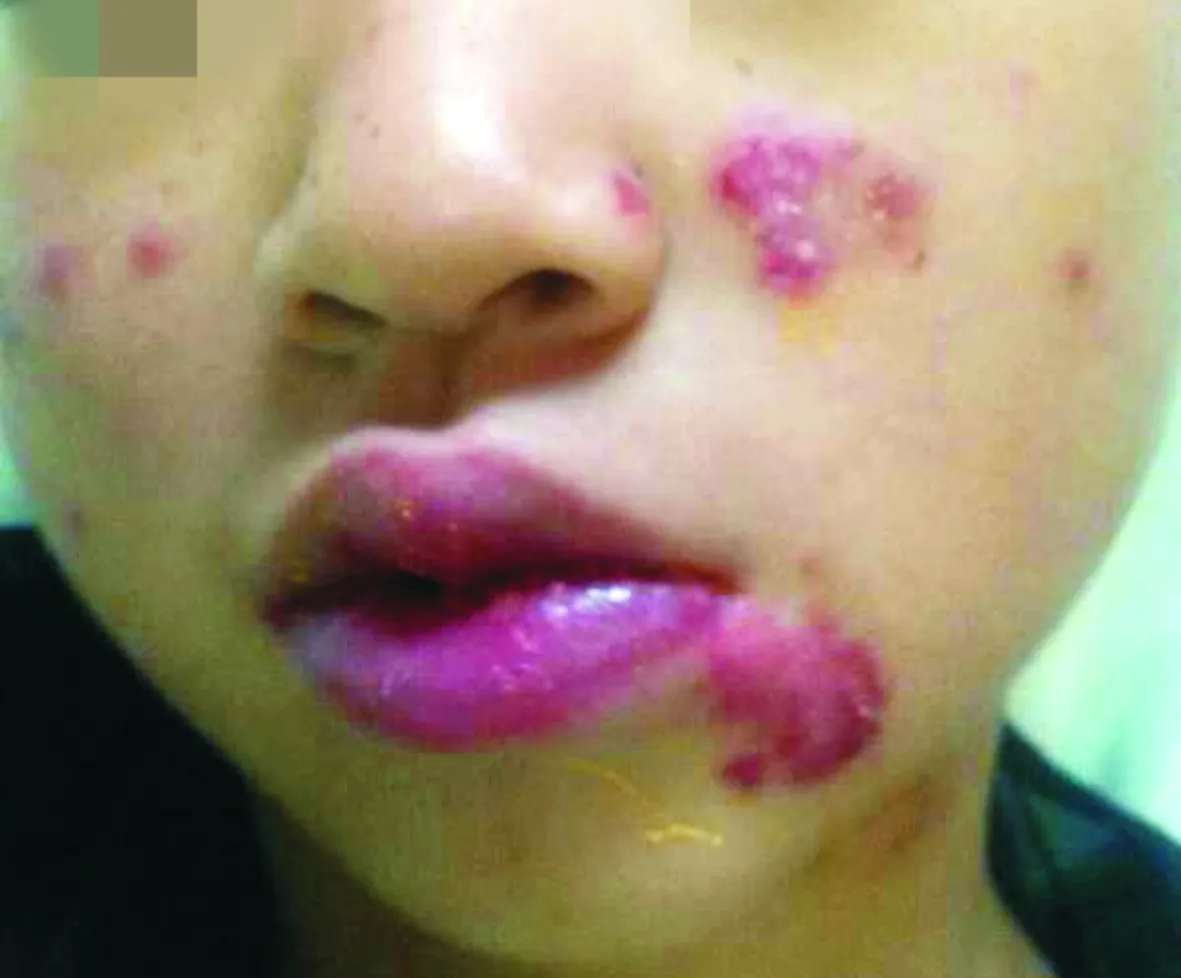
图1 本例患儿2017年7月22日第一次入院时面部特征
诊治经过:入院诊断为肝损伤,不除外噬血细胞综合征及血液系统疾病,予以保肝等对症治疗,同时加用丙种球蛋白400 mg/(kg·d),共4 d。患儿病情加重,精神状态变差,血常规三系进行性降低,最低时白细胞0.94×109/L,血红蛋白80 g/L,血小板82×109/L,铁蛋白及甘油三脂升高(见表1)。骨髓涂片可见吞噬现象(吞噬血小板及成熟红细胞)(见图2)。可溶性CD25 1 653 U/ml。TBNK亚群:CD3+T、CD4+T、CD8+T、CD19+B、CD3-CD56+NK计数均降低。诊断噬血细胞综合征,加用地塞米松抗炎,起始量为10 mg/d,逐渐减至3.5 mg/d,共6 d,患儿相关各项指标较前好转,ANA系列相关抗体由阴性转阳后逐步升高,补充诊断系统性红斑狼疮,加用羟氯喹,患儿病情逐渐好转,皮疹减轻(见图3),肝脾恢复正常,血常规、三酰甘油、乳酸脱氢酶、血沉恢复正常,好转出院。出院后继续口服羟氯喹0.1 g/d,出院1个月后复查患儿无不适但抗SM抗体、抗nRNP/Sm抗体及抗核抗体颗粒型升高。随后患儿家长自行停药6个月后皮疹较前加重,出现典型蝶形红斑样皮疹(见图4),并再次出现发热及口腔溃疡,第2次入院后抗SM、抗nRNP/Sm及颗粒型抗核抗体升高,血常规提示三系减少,进一步支持系统性红斑狼疮的诊断(SLE相关免疫学指标具体见表2),予以三轮大剂量激素冲击治疗,口服吗替麦考酚酯、羟氯喹治疗,现患儿无不适,皮疹减轻,处于随诊治疗中。
表1本例以MAS起病的SLE患儿实验室检查指标动态变化
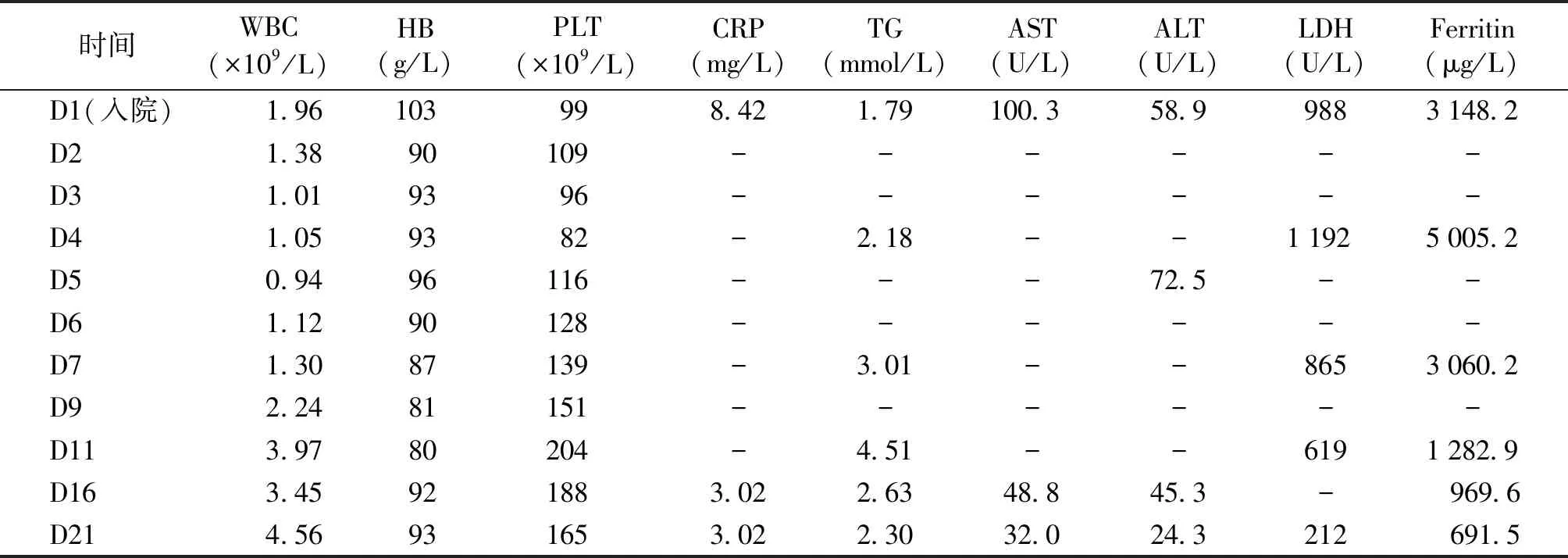
时间WBC(×109/L)HB(g/L)PLT(×109/L)CRP(mg/L)TG(mmol/L)AST(U/L)ALT(U/L)LDH(U/L)Ferritin(μg/L)D1(入院)1.96103998.421.79100.3 58.99883148.2D21.3890109------D31.019396------D41.059382-2.18--1192 5005.2D50.9496116---72.5--D61.1290128------D71.3087139-3.01--8653060.2D92.2481151------D113.9780204-4.51--6191282.9D163.45921883.022.6348.845.3- 969.6D214.56931653.022.3032.024.3212 691.5
WBC:白细胞计数;HB:血红蛋白;PLT:血小板计数;CRP:C反应蛋白;TG:三酰甘油;AST:天冬氨酸氨基转移酶;ALT:丙氨酸氨基转移酶;LDH:乳酸脱氢酶;Ferritin:铁蛋白
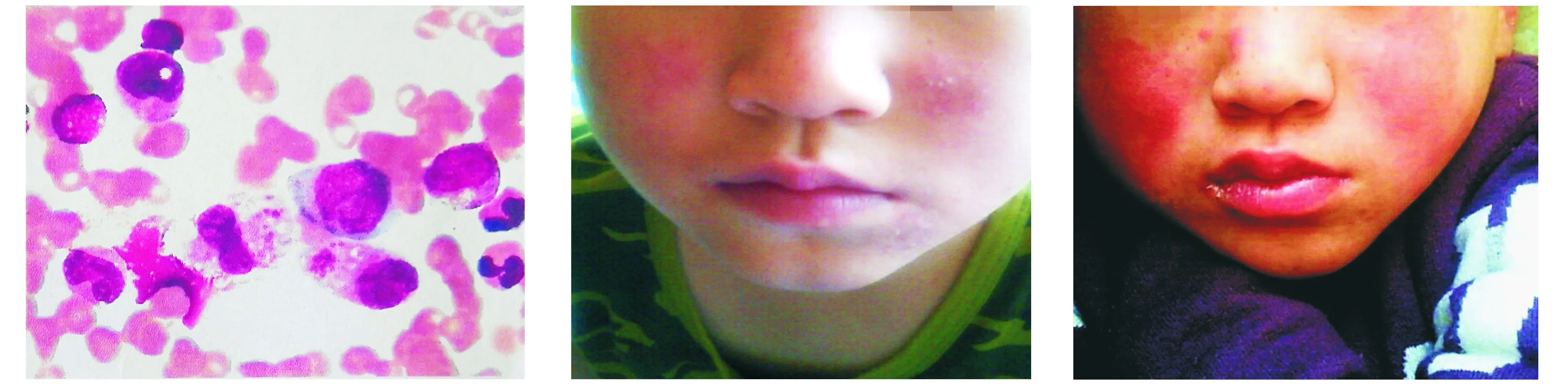
吞噬血小板及成熟红细胞图2 患儿骨髓细胞学检查可见吞噬现象 图3 患儿第一次入院出院时面部表现两侧面颊部可见明显蝶形红斑样皮疹 图4 患儿于2018年3月30日第2次入院时面部表现
表2本例以MAS起病的SLE患儿的SLE相关免疫学指标动态变化

时间抗SM抗nRNP/Sm抗核抗体颗粒型ds-DNA补体C3、C4D1-- --正常D16±+1:100-正常D21+++1∶100-正常出院1个月++++1∶320-正常第2次入院+++++1∶1000-正常
患儿行基因检测未发现与疾病表型相关的明确致病性突变,如家族性HLH的相关基因,自身炎症性的NLRC4等(患儿基因报告见图5)。我们对以MAS起病的儿童SLE进行检索,目前只检索到4例报告[3-6],其中未见明确的中文报道。本病例为第5例(这5例症状及体征见表3),其中女性4例,男性1例,年龄8-15岁,均以发热起病,其中4例存在肝脾肿大,部分患儿存在皮疹、淋巴结肿大、中枢神经系统症状、出血等表现,5例中有1例存在明确感染征象。
2 讨论
噬血细胞性淋巴组织细胞增生症(hemophagocytic lymphohistiocytosis,HLH)是由多种潜在病变引起淋巴细胞和组织细胞非恶性增生,产生细胞因子风暴所导致的一种临床综合征[7]。表现为持续发热,肝、脾肿大,全血细胞减少,皮疹,出血等,实验室检查可有脂类代谢异常、凝血障碍,骨髓发现噬血现象[8]。HLH按发病病因可分为原发性(遗传性)和继发性(反应性)两种。前者是常染色体隐性遗传,发病年龄早,80%的患者在2岁以前发病。后者在任何年龄均可发病,一般认为2岁前发病者提示原发性可能性大,8岁后发病者则提示继发性的可能性大,2-8岁发病者则根据临床表现进行判断。继发性HLH可以由免疫系统紊乱所引起,包括感染、肿瘤、自身免疫性疾病等[9]。MAS概念于1993年提出,是一种特殊类型的继发性HLH,MAS可并发于SLE,病理机制主要包括以下三种途径:①SLE患者在感染等诱发下激活了异常的免疫反应,过度分泌细胞因子(如IL-1、IL-6、INF-γ、TNF等)导致MAS;②机体产生抗造血细胞抗体,激活巨噬细胞吞噬;③免疫复合物沉积于骨髓造血细胞,激活巨噬细胞吞噬。目前对于MAS诊断主要参照2004年噬血细胞综合征诊断指南[10]。但实际中一些不典型和病情隐匿的患者在疾病早期常达不到诊断标准,因疾病进展迅速,当患者出现符合该诊断标准的症状及实验室指标,常已失去治疗的最佳时机。Parodi等[11]于2009年提出了SLE合并MAS的初步诊断指南,此指南较HLH 2004指南新增了中枢神经系统功能改变、天冬氨酸转氨酶及乳酸脱氢酶等实验室指标,血小板由≤100×109/L升高至≤150×109/L。SLE诊断参照系统性红斑狼疮国际合作组(SLICC)2009年修订的诊断标准[12]。
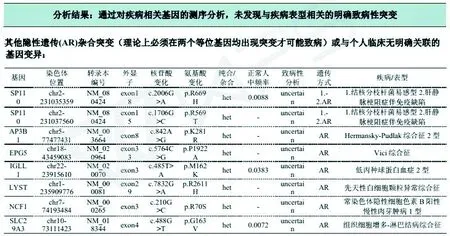
图5 本例患儿基因报告
表35例以MAS起病的SLE患儿的临床资料及临床表现
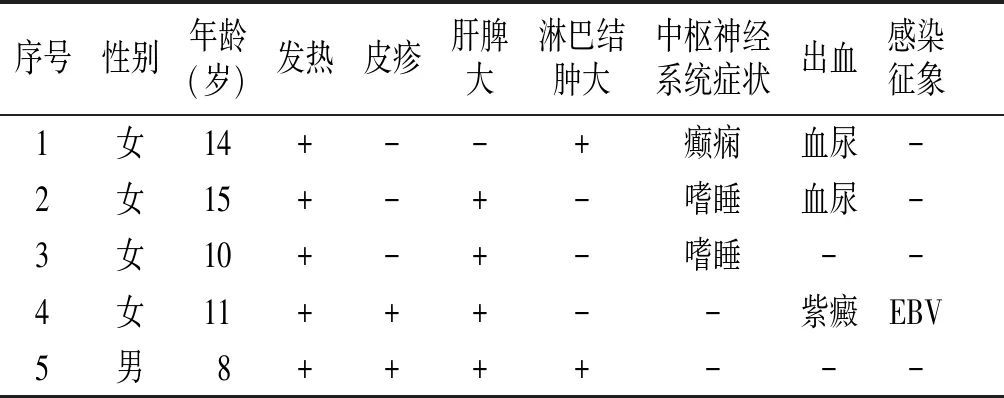
序号性别年龄(岁)发热皮疹肝脾大淋巴结肿大中枢神经系统症状出血感染征象1女14+--+癫痫血尿-2女15+-+-嗜睡血尿-3女10+-+-嗜睡--4女11+++--紫癜EBV5男8++++---
该患儿以发热起病,同时患儿具有肝脾及淋巴结肿大,血细胞三系减少,肝功能异常,三酰甘油及铁蛋白升高,骨髓涂片可见吞噬现象,符合HLH诊断。然而患儿病初参照HLH 2004诊断指南[10],存在发热、脾大、血清铁蛋白升高,尚未满足诊断标准。而依据Parodi等[11]提出的SLE合并MAS的诊断标准,患儿存在发热、二系血细胞减少(白细胞≤4.0×109/L,血小板计数≤150×109/L),天冬氨酸氨基转移酶、铁蛋白、乳酸脱氢酶等指标升高,入院时即可诊断。但该指南目前未被广泛应用,随着患儿病情进展,出现血细胞三系进行性减少,三酰甘油及铁蛋白增高,骨穿提示存在吞噬现象,诊断为MAS,诊断延迟1周,但根据患儿病史、体征及辅助检查,入院时就高度注意MAS,早期已予以针对性治疗。结合患儿发热、颜面皮疹、口腔溃疡、全血细胞减少,初诊即怀疑SLE,但ANA系列正常,补体未降低,免疫学指标未达SLE诊断标准,临床给予地塞米松及丙种球蛋白治疗,患儿热退,血细胞三系逐渐升高,肝脾淋巴结渐回缩。抗SM抗体、抗nRNP/Sm抗体及抗核抗体颗粒型由阴性转为阳性达SLE诊断标准。随后患儿ANA系列相关抗体逐渐升高并出现明显的蝶形红斑进一步支持SLE的诊断,提示MAS可能为SLE血液系统损害的表现,且部分系统性红斑狼疮患儿早期免疫学指标可能阴性[13,14]。
MAS是一种罕见且潜在致命的并发症,儿童SLE合并MAS已有较多报告,而以MAS起病的儿童SLE十分罕见。SLE合并MAS的诊断是复杂的,因为二者有很多相似的临床表现,例如发热、淋巴结肿大、神经系统症状、关节炎、皮疹、肝肿大、全血细胞减少等。MAS的特征包括高铁蛋白血症、低纤维蛋白原血症、肝功能亢进、高甘油三酯血症升高等,SLE患者全血细胞减少时应行骨髓活检排除MAS,而以MAS起病的SLE,因二者临床表现的相似性,当ANA系列相关抗体阴性时,十分容易忽略SLE。发生噬血的原因,最常见原因为感染,其中EB病毒感染最常见[15,16],而这5例病例中有4例无明确感染征象,所以当噬血的患儿无明确感染征象,且无其他明确导致噬血的原因时,我们要警惕是否合并自身免疫性相关疾病。
对于MAS的治疗,分类是关键。一类是急性狼疮噬血综合征,无明确感染征象,应用大剂量的激素冲击治疗,轻症予以泼尼松1-2 mg/(kg·d),临床表现及相关实验室检查指标好转后减量;重症采用大剂量甲泼尼龙冲击治疗, 15-30 mg/(kg·d),连续使用3-5 d后改为口服维持治疗。另一类是使用免疫抑制剂后感染相关的MAS,应予以加用抗感染药物治疗,免疫抑制剂减量。丙种球蛋白(IVIG)具有调节免疫作用,可以控制疾病的活动和感染,有助于缩短病程,在SLE相关MAS两种类型中,IVIG均可应用,一般采用1 g/(kg·d),连用2 d。除此之外,MAS其他治疗方法主要包括:①免疫抑制剂:激素治疗效果欠佳或重症MAS患者可用环孢素(CSA)静脉滴注,2-8 mg/(kg·d),分次滴注。大多患者在用药后24-48 h症状缓解,缓解后可改为4-6 mg/(kg·d)分次口服。因本药存在肝肾毒性,需根据肝肾功能调整剂量。②其他:已有使用依托泊甙、抗胸腺细胞球蛋白、TNF-α拮抗剂、IL-1受体拮抗剂/IL-6拮抗剂、血浆置换等治疗MAS的报道,但疗效尚无明确定论。③生物制剂及造血干细胞移植,由于价格昂贵,在临床中应用较为罕见,具体疗效有待进一步探究。本病例以MAS起病,发病同时即存在发热、特异性皮疹及口腔溃疡,因ANA系列阴性尚不能达到SLE诊断标准。病情演变中出现抗Sm阳性及ANA系列相关抗体滴度升高,故考虑噬血细胞综合征为SLE的血液系统损害,即急性狼疮噬血综合征。予以静点地塞米松和IVIG治疗,病情明显好转,支持临床分析。
本病例提示我们MAS可为SLE的首发症状,所以当噬血的患儿无明确感染征象时,且无其他明确导致噬血的原因时,我们要警惕是否合并自身免疫性相关疾病,早期诊断并早期给予针对性治疗。
猜你喜欢
心电与循环(2021年4期)2021-11-29
现代临床医学(2021年2期)2021-03-29
河南科学(2020年3期)2020-06-02
中国乳品工业(2019年10期)2019-12-09
中学数学杂志(2019年9期)2019-05-29
铜仁学院学报(2018年6期)2018-07-05
中国乳业(2018年8期)2018-01-25
中国眼镜科技杂志(2016年17期)2016-10-24
家庭用药(2016年7期)2016-05-14
中国继续医学教育(2015年3期)2016-01-06
