
表皮松解性掌跖角化病一家系KRT9基因突变检测
2019-03-07 13:06倪晓静周乃慧王建波
中国麻风皮肤病杂志 2019年2期
倪晓静 周乃慧 王建波 李 敏
表皮松解性掌跖角化病(epidermolytic palmoplantar keratoderma,EPPK,OMIM:144200)是一种少见的常染色体显性遗传性皮肤病,由Vörner于1901年首先描述,故又称Vörner型掌跖角化病[1]。本病主要临床表现为局限于掌跖部位的弥漫性、斑块状、有清晰红色边缘的角质增厚,表面光滑,颜色偏黄。常在出生后3个月至1年内发病[2]。Reis等首次将EPPK的致病基因定位在染色体17q12-q21上,随后在角蛋白9基因(keratin 9 gene,KRT9)中检测到一点突变R162W,确定该病的致病基因[3,4]。现报道一例EPPK,并分析其KRT9基因的突变情况。
1 先证者资料
患者,女,41岁。掌跖皮肤增厚40余年。患者出生后约4个月掌跖部开始出现弥漫性角化,随着年龄增加掌跖弥漫性角化过度明显加重,仅局限于掌跖部位,边缘清楚,色泽淡黄,表面光滑。夏季较轻,冬季干燥明显,足底常脱屑、皲裂。体格检查:一般情况好, 各系统检査未发现明显异常。皮肤科检查:掌跖部可见弥漫性角化增厚组织,淡黄色,边界清,表面干燥、质硬,双足底有少量脱屑和皲裂(图1a、b)。毛发、牙齿、甲及口腔粘膜未见明显异常。实验室检查:足底真菌镜检阴性,血、尿、粪常规及肝肾功能未见明显异常。患者拒绝行组织病理学检查。
2 家系调查
患者母亲有类似症状,父亲正常,父母非近亲结婚。家族三代共有患者6例,男2例,女4例,均为出生后3~6个月发病,皮损类似,均表现为掌跖弥漫性角化过度。目前未发病者子代均正常,发病无性别差异,无隔代遗传现象,符合常染色体显性遗传模式(图2)。
3 KRT9基因检测
3.1 实验方法 在签署书面知情同意书后,对整个家系14个家庭成员(6例患者,8名正常)进行血样采集。应用北京Tiangen公司TIANamp Blood基因组DNA提取试剂盒提取全血基因组DNA。同样方法提取100名无亲缘关系的健康对照个体基因组DNA作为对照。通过因特网(http://www.ncbi.nlm.nih.gov/)查取KRT9 mRNA序列,经BLAST比对得到该基因的基因组序列 (http://www.genome.UCSC.edu/),应用 Primer 3.0设计特异性引物对KRT9所有外显子及其侧翼序列进行PCR扩增。扩增条件:94℃预变性5 min后,94℃变性45s,56℃~60℃退火1 min,72℃延伸1 min,共35个循环。72℃延伸10 min。2%的琼脂糖凝胶电泳检查PCR产物。扩增产物纯化后直接在 ABI3730全自动DNA测序仪(美国Applied Biosystems)上进行双向测序,各测序结果采用BioEdit软件比对分析结果。用SIFT和Polyphen-2软件预测c.482A>G (p.Asn161Ser) 突变位点的有害性。
3.2 结果 PCR产物测序结果经BioEdit软件比对分析后发现,该家系6例患者的KRT9基因第482位碱基腺嘌呤(A)突变为鸟嘌呤(G),家系中8名表型正常者及100名正常对照中均未发现此突变(图3a、b)。SIFT软件对p.Asn161Ser突变的功能预测结果为有害(deleterious),其score为-4.668。Polyphen-2软件的预测结果为可能有害(probably damaging),p.Asn161Ser突变的score为1.00。提示SIFT和Polyphen-2软件预测p.Asn161Ser突变均为有害变异位点,可能影响其蛋白质功能。
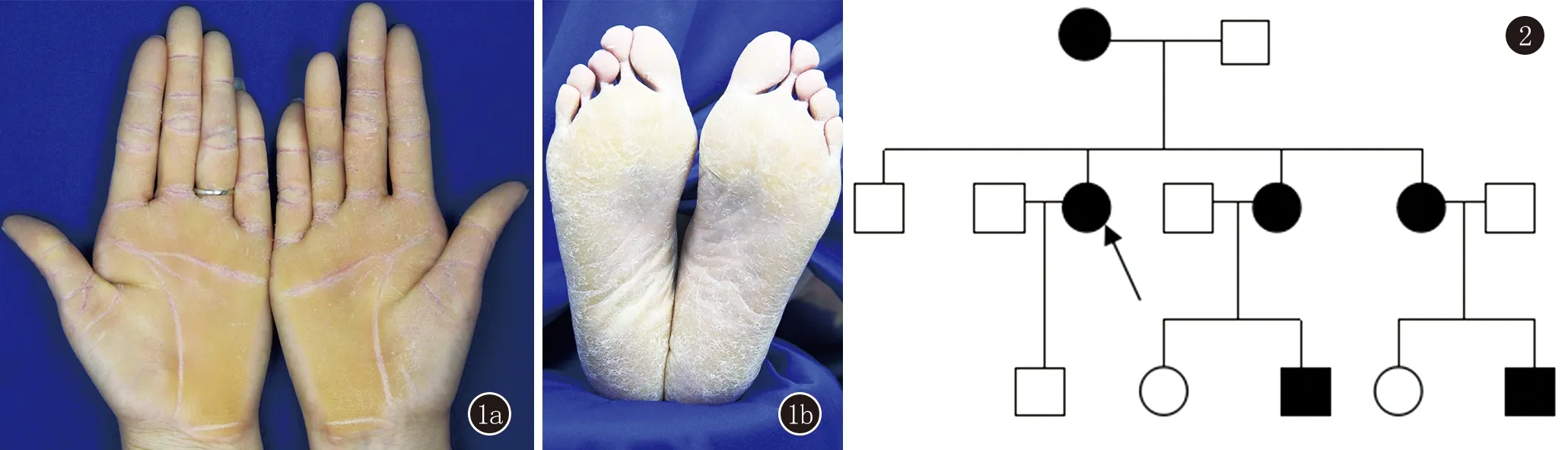
图1 a:双手掌弥漫性淡黄色角化增厚组织;b:双足掌弥漫性淡黄色角化增厚组织,上覆较多鳞屑,局部皲裂图2 家系图,箭头所指为先证者

a:正常人;b:患者,箭头示c.482A>G(p.Asn161Ser)突变
4 讨论
EPPK是一种常染色体显性遗传病,均在出生后3个月至1年内发病,通常在3~4岁表现完全,以掌跖部的皮肤弥漫性角化为主要临床特征。本病通常不累及掌跖部伸侧,部分患者伴发指节垫、甲改变(甲板增厚、浑浊)和手指畸形等临床表现,可伴发癌症、听力丧失及心力衰竭等危害严重的疾病。本研究中,先证者具有典型的临床表现,结合其家系符合常染色体显性遗传模式,EPPK基本可诊断,基因突变分析进一步明确诊断。
近些年的遗传及分子学研究表明,EPPK是由编码角蛋白9的基因KRT9发生突变所致。KRT9是一种Ⅰ型角蛋白,仅表达于掌跖部表皮的棘突层。角蛋白基因的突变常会引起各种表皮和非表皮的细胞脆性问题[5,6]。突变的角蛋白纤维削弱了细胞骨架,当细胞遭受物理创伤时,导致角质形成细胞的结构网络崩溃。KRT9基因突变热区位于杆状功能域lA区前端的15个氨基酸和2B区末端的10~11个氨基酸,这两个区域是两段在进化上高度保守的氨基酸序列:螺旋起始基序和螺旋终末基序[7,8]。迄今,在不同的EPPK家系中检测到至少32种不同的KRT9基因突变位点,其中25种发生在1A区,6种发生在2B区[9-12]。2013年,Fuchs-Telem等[13]报道一新发突变p.[Leu11Val; Leu11_Gln172del],该突变位点除了涉及1A区,还累及KRT9非螺旋区的头部,打破了学者们常规认为KRT9基因突变位点通常在1A和2B区域的观念。KRT9基因1A区的突变p.R163W是EPPK中最常见的突变类型,约占31.71%,在我国的发生频率约为31.03%[9]。目前EPPK仍没有明确的基因型-表型相关性分析,可见本病的发病因素或许比人类迄今所了解的更复杂。
本家系中患者的KRT9基因第482位碱基腺嘌呤(A)突变为鸟嘌呤(G),导致1号外显子中第161位密码子由AAT→AGT,即正常的天冬酰胺(Asparagine,Asn)由丝氨酸(Serine,Ser)取代,即c.482A>G(p.Asn161Ser)。此突变位点既往国内有报道[14,15],当时报道为“KRT9基因第1外显子第160位密码子发生AAT→AGT的突变,导致第160位的天门冬氨酸被丝氨酸取代(N160S)”。新版本的KRT9基因序列发表于2005年4月23日,突变R162W更换为R163W[gi:55956898],故此突变位点最新的标注为c.482A>G(p.Asn161Ser)。此外,既往文中的天门冬氨酸应更改为天冬酰胺。本家系突变位点c.482A>G(p.Asn161Ser)处于杆状功能域螺旋起始基序,杆状结构域为α-螺旋二级结构,突变后可能引起氨基酸理化性质的改变,造成角蛋白亚单位受损,从而破坏角蛋白中间丝聚合的稳定性,最终导致本家系患者掌跖角化过度的发生。另外 SIFT和Polyphen-2软件也同时预测c.482A>G(p.Asn161Ser)突变为有害变异位点,进一步印证该突变可能影响蛋白质功能。此突变位点也为热点突变,目前所报道的均发生在亚洲国家,日本、韩国及国内均有家系报道[6]。国内报道的家系中患者有伴发指节垫、甲改变和手指畸形等临床表现[14,15]。本家系患者中均无以上病变,以上病变是否与KRT9基因突变有关及其致病机理如何还有待进一步研究。
总之,EPPK根据其临床表现结合基因突变分析即可确诊,但具体的发病机制尚不明确。目前治疗方法有局部对症治疗、口服维A酸类药物、CO2激光疗法及分层皮片移植术等,但疗效均不佳,本病理论上仍寄希望于基因治疗。目前的病例报道、基因测序及功能研究等进一步为该病的基因诊断、遗传咨询和基因治疗提供理论基础。
猜你喜欢
纺织报告(2022年5期)2022-11-22
中国畜牧杂志(2022年1期)2022-11-06
天津医科大学学报(2021年4期)2021-08-21
数学学习与研究(2021年18期)2021-08-06
皮肤病与性病(2021年3期)2021-07-30
健康之家(2021年19期)2021-05-23
毛纺科技(2021年3期)2021-04-06
中国麻风皮肤病杂志(2021年2期)2021-01-02
东方教育(2017年14期)2017-09-25
课程教育研究·学法教法研究(2016年1期)2016-03-17
