
石墨相氮化碳负载磷钨酸杂化材料的制备及其氧化脱硫催化性能
2019-03-06 02:33,,,,*,,*,,
燃料化学学报 2019年2期
, , , ,*, ,*, ,
(1.辽宁石油化工大学 化学化工与环境学部, 辽宁 抚顺 113001; 2.中国石油石油化工研究院 大庆化工研究中心, 黑龙江 大庆 163714)
随着人们的消费水平大幅提高,车辆数量也明显增加,导致汽柴油的需求增加[1]。原油中的硫氧化物对于发动机和环境造成的危害不可忽视。世界各国都相继出台了更加严格的燃料油标准,对车用燃料油的深度脱硫加工工艺提出了挑战。中国汽柴油的国VI标准对硫含量的要求为10 μg/g,而目前的应用最为广泛的加氢脱硫工艺很难生产出满足要求的车用燃料油。研究者开始寻求操作条件温和,具有深度脱硫效果的非加氢脱硫技术,主要包括萃取脱硫[2]、生物脱硫[3]、络合脱硫[4]、吸附脱硫[5,6]和氧化脱硫[7,8]等。其中,氧化脱硫技术反应条件比较温和,而且加氢脱硫活性低的大分子噻吩类组分,却容易被氧化生成高极性的噻吩砜,然后通过萃取或吸附的方法从油品中脱除。
过氧化氢是氧化脱硫技术中最常用的氧化剂之一[9-11],主要原因是将其用于氧化反应后的产物是水,符合“绿色化学”的要求。但是,采用过氧化氢作为氧化剂会产生界面传质的问题。过氧化氢存在于水相,而有机含硫化合物存在于油相,所以氧化反应必须要克服水-油两相的界面限制。针对水-油界面传质的问题,目前主要有引入第三溶剂[12]、加入表面活性剂[13]和建立微乳液体系[14]三种解决途径。然而,离子液体价格昂贵,萃取剂的使用会带来环境问题,微乳液体系存在一定的使用局限性,所以如能解决以过氧化氢为氧化剂的脱硫工艺中界面传质问题,将极大地推动该技术的工业化发展。
目前,在能够催化过氧化氢氧化含硫化合物的催化剂之中,杂多酸的结构稳定,对环境无污染,其中,具有Keggin结构的12-磷钨酸及其盐类是最有应用前景的活性组分,被广泛应用于氧化脱硫反应中[7,10,15]。另一方面,制备负载型的杂多酸类催化剂是解决由于比表面积低而活性不高的有效途径。在众多的载体中,石墨相氮化碳作为新兴的催化材料被广泛的应用,因其独特的类石墨层状堆积结构和sp2杂化的π共轭电子能带结构,使其具有多种优异的物理和化学性质,加之其制备简单、原料廉价、无毒,且具有良好的热稳定性和化学稳定性等性质。邢鹏飞等[16]以三聚氰胺为原料,焙烧生成g-C3N4,再将g-C3N4同MoO3混合后经焙烧制备得到MoO3/g-C3N4催化剂,实验结果表明,反应温度为70 ℃、反应时间60 min时的反应条件下脱硫率可以达到94.8%。李秀萍等[17]合成了NiWO4/g-C3N4催化剂,以1-丁基-3-甲基咪唑四氟硼酸盐离子液体为萃取剂,双氧水为氧化剂,考察了该催化剂对模拟油的催化脱硫性能,结果表明:在反应温度80 ℃、反应时间140 min的反应条件下,脱硫率为97.35%,该循环体系重复五次脱硫率仍能达到95.47%。Li等[18]采用微波辅助自组装法制备了CeO2/attapulgite/g-C3N4催化剂,以双氧水为氧化剂考察了催化氧化脱硫反应性能。由于未能很好解决水-油界面传质问题,该体系只能适用于脱除初始含硫化合物浓度为200 μg/g的油品。在众多报道的杂多酸(盐)与H2O2组成的氧化体系中,由于水-油界面传质的限制,会产生氧化剂的加入量大、环境不友好和不能处理硫含量高的油品等问题。
以1-丁基-3-甲基咪唑溴离子液体和磷钨酸为原料,合成了一种具有Keggin结构的杂多酸杂化材料BPWO,并通过原位沉淀法将BPWO负载到2D结构的多孔g-C3N4上,得到复合催化剂BPWO/g-C3N4。以双氧水为氧化剂,选择DBT的正庚烷溶液为模拟油品,考察该催化剂在间歇釜反应器对DBT的氧化性能,优化了BPWO负载量、催化剂添加量、O/S物质的量比和反应温度等反应条件,并对催化氧化脱硫过程中的机理做了初步探讨。新型的BPWO/g-C3N4催化剂具有双亲性,可以在反应过程中同时吸附油相中的DBT和水相中的H2O2,加强水-油两相界面的传质,且载体g-C3N4提高了BPWO的分散度,增大了活性位点与DBT和双氧水接触面积,进而提高了催化剂的氧化性能。
1 实验部分
1.1 实验仪器与药品
磷钨酸、双氧水(30%)、1-丁基-3-甲基咪唑溴离子液体、尿素、正庚烷和无水乙醇,均为分析纯,采购于国药集团化学试剂有限公司。蒸馏水和DBT(99.9%)自制。
磁力搅拌器(HJ-4,江苏省金坛市荣华仪器制造有限公司);循环水式真空泵(SHZ-D,巩义市予华仪器有限公司);电子天平(WT-B1003,杭州万特衡器有限公司);离心机(800型,金坛市荣华仪器制造有限公司);台式干燥箱(101-1 EBS,北京市永光明医疗仪器有限公司);恒温水浴锅(DF-101S,巩义市予华仪器有限责任公司);气相色谱仪(6890 plus,Agilent);马弗炉(SX2-4-10,沈阳市工业电炉厂)。
1.2 催化剂的制备
杂多酸杂化材料BPWO的制备方法参照文献[19],取1 g [Bmim]Br离子液体溶于80 mL去离子水形成溶液A,取4.38 g磷钨酸溶于50 mL去离子水中形成溶液B,在搅拌的条件下,将溶液B逐滴加入溶液A中得到白色沉淀,继续搅拌1 h,所得沉淀采用去离子水洗涤,用AgNO3检测Br-是否洗涤干净,将洗涤后的沉淀置于80 ℃环境下干燥8 h,研磨即可得到BPWO。
g-C3N4的制备方法如下:将尿素置于坩埚中,然后将其放入马弗炉焙烧,升温程序:初始温度30 ℃,以5 ℃/min的升温速率升到550 ℃,550 ℃保持2 h。
负载型BPWO/g-C3N4催化剂的制备方法如下:取2 g载体g-C3N4分散在100 mL去离子水中,并加入0.100-1.596 g的[Bmim]Br离子液体,充分搅拌得到悬浊液A,然后将0.437-6.993 g磷钨酸溶于30 mL的去离子水,充分溶解形成溶液B,在磁力搅拌的条件下,将溶液B滴加到悬浊液A中得到淡黄色的沉淀,再采用去离子水洗涤,并用AgNO3检测Br-是否洗涤干净,将洗涤后的沉淀置于80 ℃环境下干燥8 h,研磨得到不同负载量的负载型BPWO/g-C3N4催化剂。该过程可以描述如下:

(1)
1.3 催化剂的表征
采用日本岛津公司的XRD-7000型X射线衍射仪对样品进行物相的长程有序度分析,CuKα辐射源,管电流30 mA,管电压40 kV,Ni滤波片,扫描10°-80°;采用FT-IR-800型红外光谱仪对样品进行物相的短程有序度分析,KBr压片,室温下测试;采用日本JASCA公司的UV-550型紫外-可见光谱仪(UV-vis)对自制样品进行结构分析。采用美国麦克仪器公司的Micrometrics2010型物理吸附仪测定样品的比表面积;采用JEOL JEM2010型透射电子显微镜(TEM)对样品进行显微结构观测。采用赛默飞世尔科技有限公司Thermo ESCALAB 250型光电子能谱仪对样品进行元素分析。
1.4 催化剂氧化性能的评价
以DBT的质量百分含量为800 μg/g的正庚烷溶液为模拟油品,采用配有冷凝回流的间歇釜式反应器,以双氧水作为氧化剂,考察BPWO/g-C3N4催化剂对模拟油品中DBT的氧化性能。具体过程描述如下:向50 mL三口瓶中加入25 mL模拟油品,并在磁力搅拌条件下,加入一定量的催化剂,采用恒温水浴控制反应温度,按照相应的O/S物质的量比加入双氧水,每隔30 min取样分析。原料和样品采用Agilent 6890 plus型气相色谱仪进行分析,HP-5毛细管色谱柱(氢火焰检测器,FID),氮气为载气,初始温度120 ℃,恒温3 min,15 ℃/min升至260 ℃,保持5 min。
2 结果与讨论
2.1 表征与分析
2.1.1XRD表征
图1为g-C3N4、BPWO和BPWO (20%-80%)/g-C3N4样品的XRD谱图。由图1可知,BPWO样品的谱图在7°-11°、17°-23°、25°-28°和33°-38°出现了四组非常明显的衍射峰,与H3PW12O40的XRD谱图中Keggin型杂多酸的特征峰高度一致,说明所合成的BPWO具有Keggin型杂多阴离子的结构特征[20]。g-C3N4样品的谱图显示两个特征峰,分别位于13.1°和27.5°处,分别对应g-C3N4的(100)和(002)晶面(JCPDS 87-1526)[21]。BPWO (20%-80%)/g-C3N4样品的谱图显示了Keggin型杂多阴离子结构和g-C3N4的特征衍射峰,且BPWO的特征峰强度随着负载量的增加而增强。 BPWO (20%-60%)/g-C3N4样品的BPWO的特征峰强度很接近,说明活性组分BPWO被均匀地分散到了载体g-C3N4上[17],有助于提高催化剂的催化活性。

图 1 g-C3N4、BPWO (20%-80%)/g-C3N4和BPWO样品的XRD谱图
2.1.2FT-IR表征
图2为g-C3N4、BPWO和BPWO (20%-80%)/g-C3N4样品的FT-IR谱图。

图 2 g-C3N4、BPWO (20%-80%)/g-C3N4和BPWO样品的FT-IR谱图
在BPWO红外谱图中,1080、980、897和803 cm-1分别归属为Keggin结构中V(P-O)、V(W=O)、V(W-Oc-W)和V(W-Oe-W)的振动吸收[22],证明所合成的BPWO催化剂保持了Keggin结构特征。在g-C3N4的红外光谱谱图中,吸收峰主要集中在三个区域:808 、1240-1650 和3100-3500 cm-1。808 cm-1附近的吸收峰对应三嗪环状化合物的弯曲振动特征吸收[23],1240-1650 cm-1的吸收峰对应碳氮芳香杂环化合物的伸缩振动特征吸收[24]。3100-3500 cm-1的宽波段对应N-H键的伸缩振动吸收,说明g-C3N4层状结构的边缘存在N-H键[25]。由图2可知,随着BPWO负载量的增加,除了保持了g-C3N4的全部特征峰以外,位于980 cm-1处的W=O振动吸收清晰可见,说明所合成的BPWO/g-C3N4同时具有Keggin型磷钨酸和g-C3N4的结构特征。
2.1.3UV-vis表征
图3为g-C3N4、BPWO和BPWO (20%)/g-C3N4样品的UV-vis谱图。从g-C3N4的谱图中可以看出,吸收边界约为452 nm,与文献报道结果一致[26]。在BPWO的谱图上,200-400 nm出现的吸收带,是杂多酸Keggin结构中氧原子对中心原子的电荷转移所形成[27],其中,在212 nm处的吸收带是由于Keggin结构中O原子到P原子的电荷转移,257和325 nm处的吸收带可以归属为Keggin结构中配体原子O2-到W6+的电荷转移[28]。在BPWO (20%)/g-C3N4的谱图上可以看出,负载型催化剂BPWO (20%)/g-C3N4的吸收带与BPWO相同,说明所合成的BPWO/g-C3N4同时具有Keggin型磷钨酸和g-C3N4的结构特征。

图 3 g-C3N4、BPWO和BPWO(20%)/g-C3N4样品的UV-vis谱图
2.1.4TEM表征
图4为g-C3N4、BPWO和BPWO (20%)/g-C3N4样品的TEM照片。
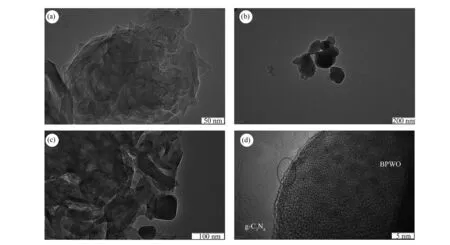
图 4 g-C3N4(a)、BPWO(b)和BPWO (20%)/g-C3N4((c)、(d))样品的TEM照片
图4(a)为g-C3N4的TEM照片,可以看出经NaClO处理后,g-C3N4呈现出几乎透明的2D层状结构。不仅如此,大小不一的多级孔结构在g-C3N4上清晰可见。图4(b)为BPWO的TEM照片,可以看出BPWO为无规则形貌的纳米颗粒。粒径约为100 nm,且团聚现象较严重。图4(c)为BPWO (20%)/g-C3N4的TEM照片,可以看出BPWO成功负载在g-C3N4表面,且分散性良好。将图4(c)中选定的两催化剂组分的交界面经过多次放大,结果图4(d)。在界面处,两组分的晶格条纹清晰可见。
2.1.5XPS表征
图5为BPWO和BPWO (20%)/g-C3N4样品的XPS谱图。由图5(a)和5(b)可知,BPWO (20%)/g-C3N4具有C、N、O、P和W五种元素组成,再次说明BPWO负载到了g-C3N4上。图5(c)的C 1s谱图可分为两个峰,其中,284.6 eV处的峰归属为环状结构中sp2杂化的C原子(N-C=N),287.8 eV的峰归属为sp3杂化的C原子(C-(N)3)[29-31]。图5(d)中N 1s能级的398.3 eV的峰归属为sp2杂化的N原子(C-N=C)[32]。图5(f)中BPWO (20%)/g-C3N4中的W 4f7/2和Wf5/2的特征峰分别位于35.25和37.5 eV[33]。
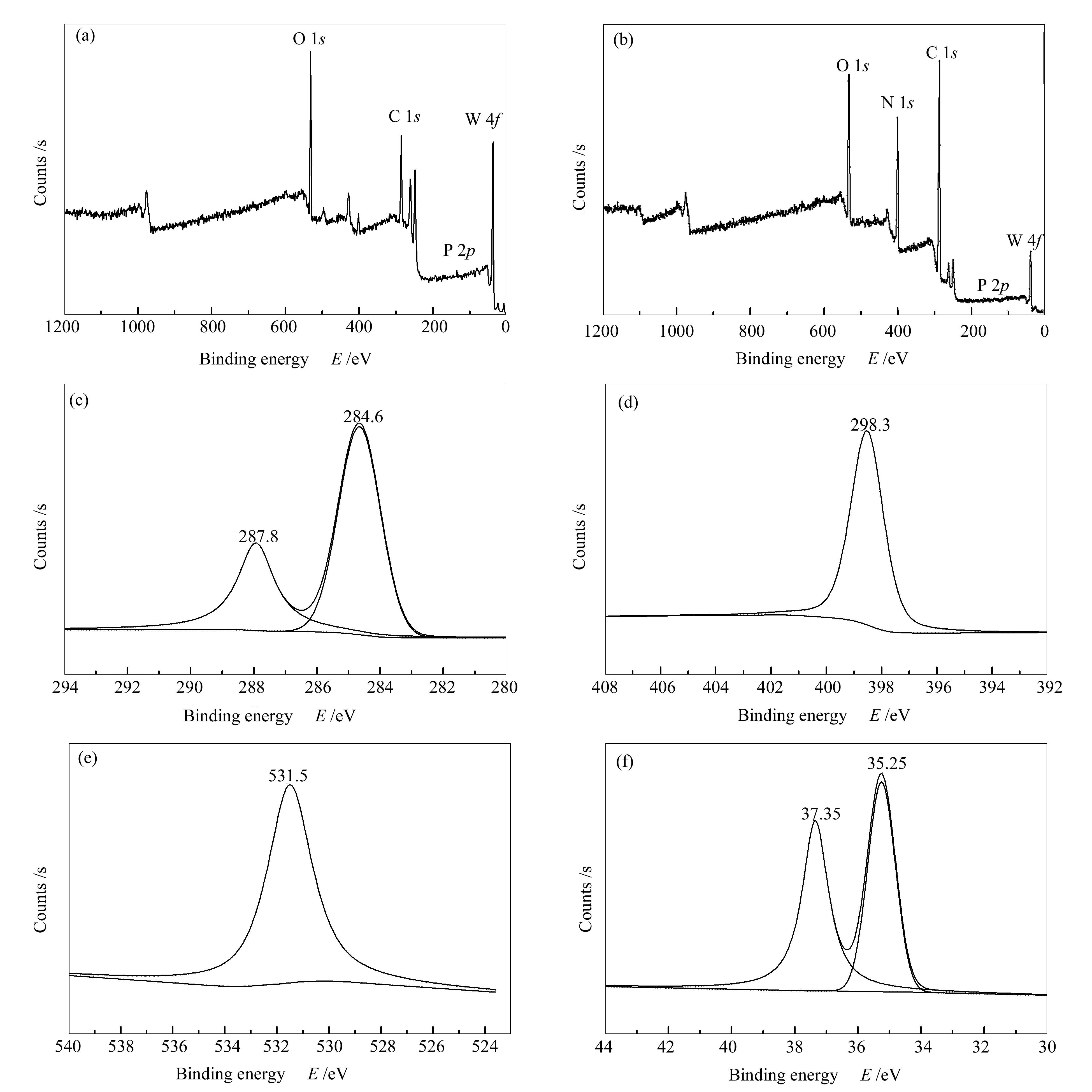
图 5 BPWO和BPWO(20%)/g-C3N4的XPS谱图
2.2 氧化脱硫条件考察和分析
2.2.1不同催化剂氧化DBT活性对比
表1为BPWO负载量对DBT的氧化活性的影响。由表1可知,纯载体g-C3N4对DBT没有氧化活性,DBT的转化率为2.3%可能是由于物理吸附的结果;非负载的BPWO对DBT表现出较低的氧化活性,在反应180 min时,DBT的转化率为14%;当将BPWO负载在g-C3N4上制备负载型的催化剂后,其氧化活性有明显升高,并在负载量为20%时出现了最大值,DBT的转化率为87%,继续增加BPWO的负载量,DBT在催化剂上的转化率开始下降,从样品的比表面积数据分析,可以推测活性的下降是由于催化剂比表面积减小而造成的。因此,选择负载量为20%的催化剂考察其他条件对DBT氧化反应的影响。

表 1 不同催化剂的比表面积及其氧化DBT的活性
reaction conditions: 40 ℃, 180 min, the amount of catalyst was 0.05 g, and with an O/S molar ratio of 4.0
2.2.2催化剂加入量对DBT转化率的影响
图6为催化剂的加入量对DBT氧化反应的影响,由图6可知,在相同的反应条件下,当体系无催化剂时,DBT转化率几乎为零,当催化剂质量从0.025 g提高到0.075 g时,DBT的转化率有不同程度的提高,增加催化剂加入量为反应提供更多的活性中心,有利于促进DBT的氧化反应。综合考虑之后,选择催化剂的加入量为0.05 g考察其他条件对DBT氧化反应的影响。
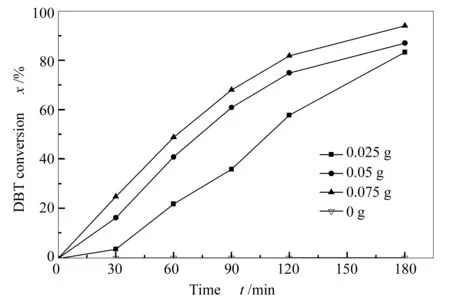
图 6 催化剂加入量对DBT在BPWO/g-C3N4-H2O2体系中转化率的影响
2.2.3反应温度对DBT转化率的影响
图7为反应温度对DBT氧化反应的性能影响。
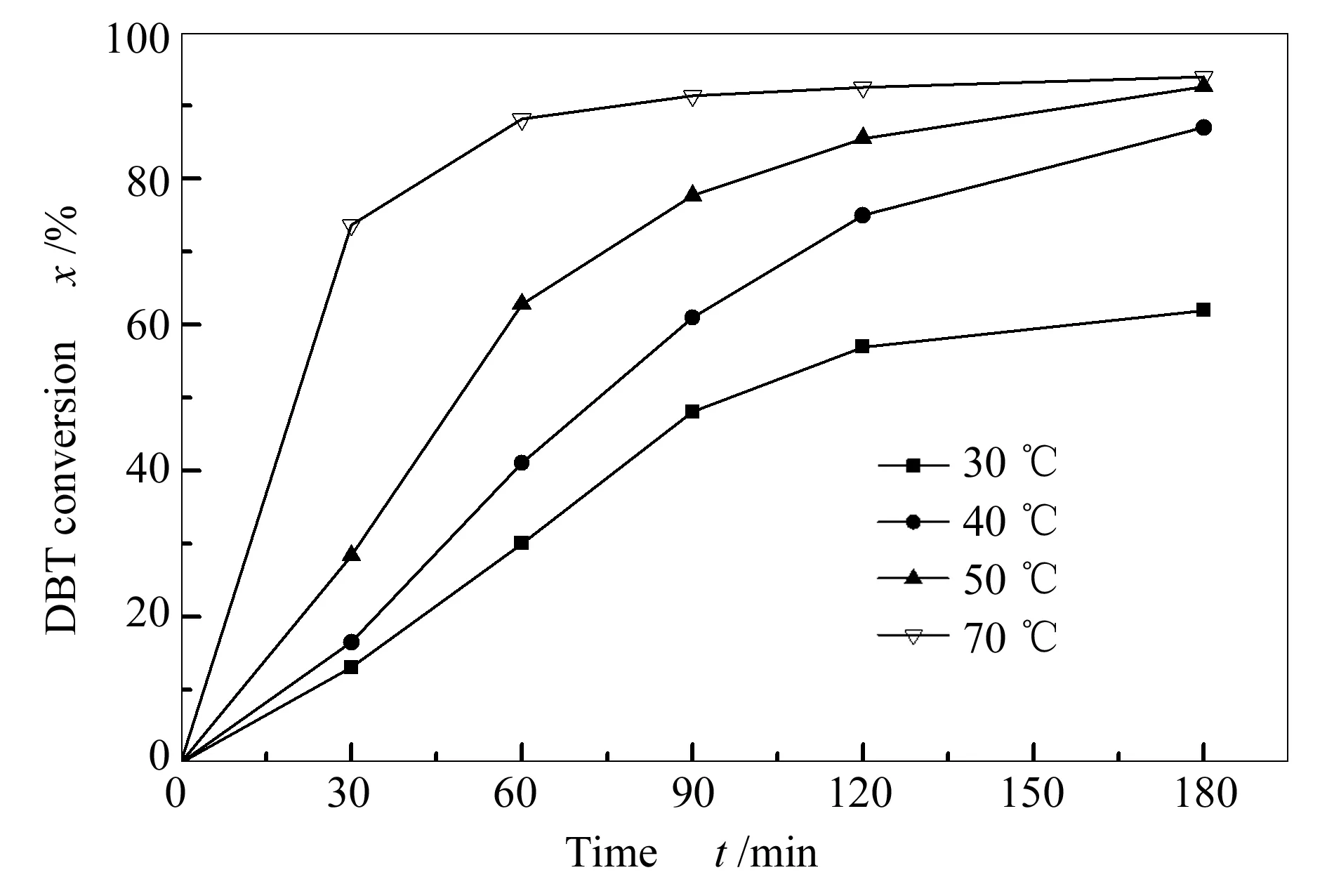
图 7 温度对DBT在BPWO/g-C3N4-H2O2体系中转化率的影响
由图7可知,DBT在BPWO (20%)/g-C3N4催化剂上的转化率随着反应温度的升高而增大,在40 ℃,反应时间180 min,DBT的转化率为87%,当反应温度升高至50 ℃时,DBT的转化率可以达到92.61%,进一步提高反应温度到70 ℃,DBT转化率提高的不明显。在氧化DBT的反应过程中,存在两个与H2O2有关的平行反应,分别是H2O2的热分解和DBT的氧化。从热力学角度分析,提高温度有助于促进这两个反应。综合考虑之后,将反应的温度设定在50 ℃考察其他条件对DBT氧化反应的影响。
2.2.4O/S物质的量比对DBT转化率的影响
图8考察了氧化剂的加入量对DBT氧化反应的影响。由图8可知,在不加氧化剂的条件下,DBT的转化率为2.38%,体系中加入过氧化氢之后,DBT的转化率明显提高,在O/S=2.0和O/S=4.0的条件下(均为物质的量比),反应时间180 min时,对DBT的转化率分别为56.8%和92.6%。进一步提高氧化剂的量到O/S=6.0,反应180 min时,DBT被完全氧化。综合考虑之后,选择O/S=4.0考察其他条件对DBT氧化反应的影响。

图 8 O/S物质的量比对DBT在BPWO/g-C3N4-H2O2体系中转化率的影响
2.2.5催化剂循环使用性能考察
图9为BPWO(20%)/g-C3N4催化剂重复使用性能。
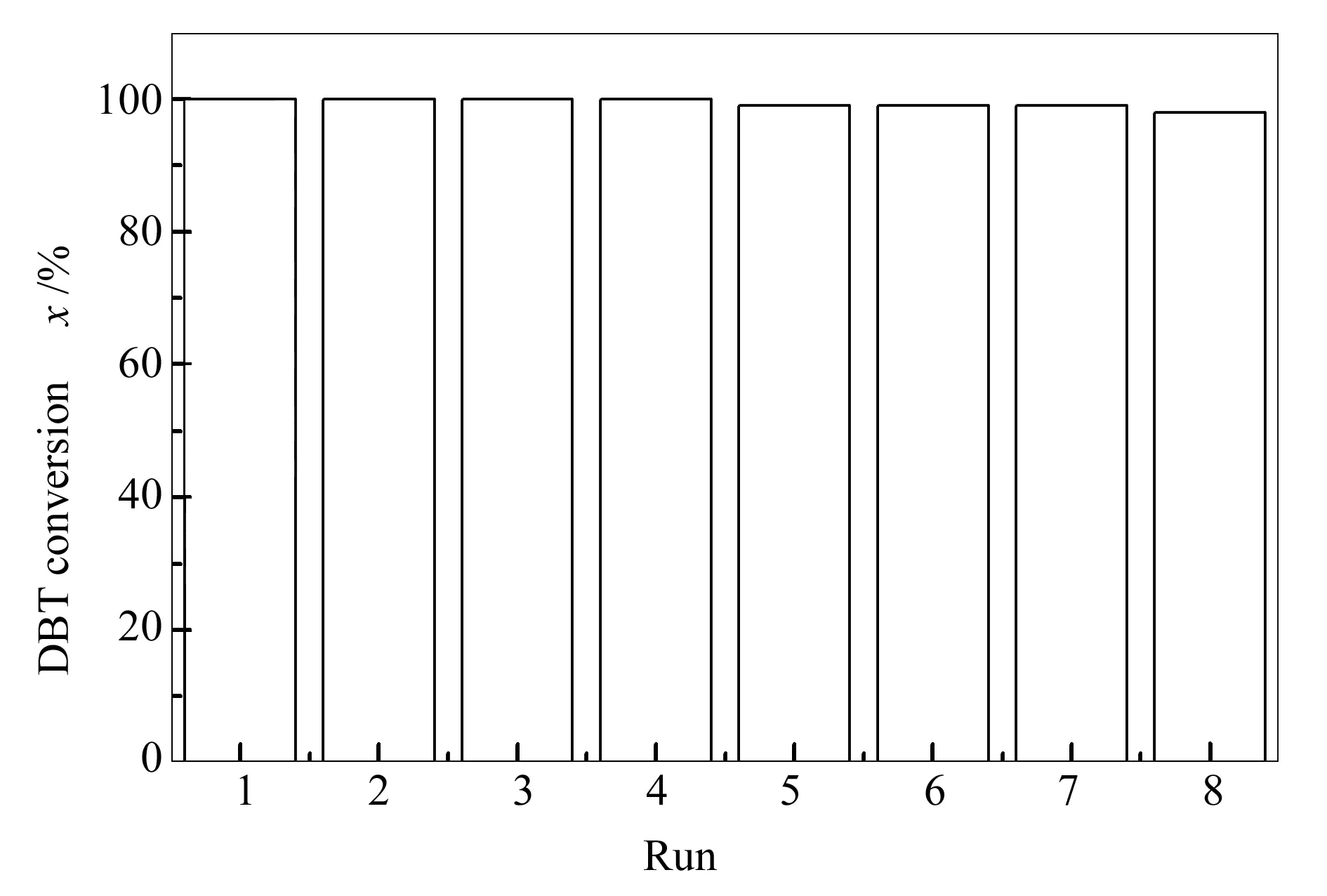
图 9 BPWO (20%)/g-C3N4催化剂的循环使用性能
反应过程描述如下:反应温度50 ℃,O/S物质的量比为6.0,反应时间180 min。在氧化反应之后,采用离心法分离催化剂,并将回收的催化剂在80 ℃干燥8 h除去催化剂吸附的H2O和未反应的H2O2之后,再用于下一次反应。
由图9可知,BPWO (20%)/g-C3N4具有优异的重复使用性能,可以循环使用至少八次而对DBT的氧化活性没有明显降低,而DBT转化率下降的原因,可能是由于催化剂在回收的过程中所造成的质量损失而导致。
2.3 催化氧化反应机理
图10为BPWO/g-C3N4催化氧化脱硫机理图。在此氧化体系中,由BPWO/g-C3N4、H2O2和模拟油形成了固-液-液三相。[Bmim]-基团的引入,使复合型催化剂BPWO/g-C3N4具有了亲油和亲水的双亲性质,有效地解决了水-油界面传质问题。首先,g-C3N4负载的[PW10O40]3-先被H2O2氧化为{PO4[WO(O2)2]4}3-。随后DBT被{PO4[WO(O2)2]4}3-氧化为二苯并噻吩亚砜和二苯并噻吩砜[34-37]。

图 10 BPWO/g-C3N4催化氧化脱硫机理图
3 结 论
本实验制备出的g-C3N4具有多级孔结构,BPWO被均匀分散到了g-C3N4上。所制备的负载型BPWO/g-C3N4催化剂具有咪唑阳离子基团和Keggin型杂多阴离子基团的结构特征。
负载型BPWO/g-C3N4催化剂对DBT具有较高的氧化活性。催化剂最佳的负载量为20%,在50 ℃和O/S物质的量比为6.0的条件下,反应180 min,即可完全氧化模拟油品中800 μg/g的DBT。催化剂具有优异的重复使用性能,经离心分离和干燥处理之后,可以循环使用至少八次而对DBT的氧化性能没有没有明显降低。
猜你喜欢
江西农业学报(2022年8期)2022-11-04
河南科技(2022年8期)2022-05-31
烟台果树(2021年3期)2021-07-21
中学化学(2019年4期)2019-08-06
中学化学(2019年4期)2019-08-06
农业与技术(2017年24期)2018-01-19
山西果树(2017年3期)2017-05-31
中学化学(2015年2期)2015-06-05
新课程·中学(2014年7期)2014-10-24
理科考试研究·高中(2014年8期)2014-10-17
