
煤燃烧过程中砷与氮氧化物的反应机理
2019-03-06 02:33,,
燃料化学学报 2019年2期
, ,
(华北电力大学 能源动力与机械工程学院, 河北 保定 071003)
煤炭是中国的主要能源,燃煤造成的环境污染问题被高度重视[1]。煤粉燃烧过程中不仅产生大量的常规污染物(SOx、NOx、颗粒物等),煤中一些痕量元素也会迁移释放到大气中[2]。这些痕量元素包括汞、砷、铅等重金属,其中,由于砷具有极强的毒性和致癌性[3,4]而备受关注。
一般认为,煤燃烧过程中砷首先从碳颗粒内部挥发到烟气中,烟气从炉膛出口到尾部烟道流动过程中温度不断降低,烟气中的砷逐渐由气相向固相转变,发生形态和价态的迁移转化[5-7]。形态不同的砷,对环境的危害性也不同。因此,研究煤燃烧过程砷的形态变化,有助于揭示砷的迁移转化特性,对砷的排放控制提供一定的理论依据。Contreras等[8]通过HSC Chemistry模拟煤燃烧中砷的形态变化,发现在温度高于1073 K时,砷主要以AsO2(g)和AsO(g)存在。刘迎晖等[9]采用化学热平衡软件FACT分析了还原性和氧化性气氛煤燃烧砷形态的转变,结果表明,在还原性气氛下,700-800 K时,出现了气相单质砷,温度进一步升高后,砷的主要产物为AsO(g);氧化性气氛下,高于800 K时,主要产物为AsO(g)。Urban等[10]利用高斯软件在不同的方法/基组条件下计算并对比了As与氧气的反应的能量。Monahan-Pendergast等[11]对大气条件下As与一些自由基(OH、HO2等)的反应机理进行了研究。Urban等[12]利用密度泛函理论和从头计算方法研究了燃煤烟气中As与HCl之间的反应动力学。
煤燃烧过程中,烟气中含有大量的氮氧化物,如N2O、NO2和NO等。很多学者对氮氧化物的释放特性进行了研究[13-15],发现氮氧化物能与其他气体或焦炭发生氧化还原反应。因此,烟气中的氮氧化物可能与气相砷发生反应,但这方面的研究报道极少。限于目前的测量水平,很难对燃烧过程中砷的释放机理进行直接测定,并且燃烧过程中释放的砷含量较少,化学反应时间极短,利用实验也很难精确地确定其反应机理。量子化学计算是一种计算分子几何结构和反应机理的有效手段[16,17],在量子力学的理论框架下用计算机模拟化学反应过程,并通过计算出动力学和热力学参数,为深入研究燃煤过程中砷的释放与迁移提供理论依据。
本研究选取了燃烧过程中三种氮氧化物(N2O、NO2和NO),应用密度泛函理论研究了其与砷之间的反应性能。
1 计算理论
1.1 理论方法和基组
量子化学方法是一种精准计算分子构型及能量的理论方法,而在量子化学计算中密度泛函得到了广泛的应用。本研究采用密度泛函理论中的B3LYP方法,在6-311G*基组水平上研究煤燃烧过程中砷与含氧气体反应的微观机理,优化得到的反应物(Reactant,简写为R)、过渡态(Transition State,简写为TS)、中间体(Intermediate,简写为M)和产物(Product,简写为P)的构型,并通过频率计算的结果验证各结构的真实性并得到零点矫正能(ZPE)。过渡态有且只有一个虚频,且虚频的振动方向指向反应方向,并通过内禀反应坐标IRC(Intrinsic reaction coordinate)计算验证反应物、中间体、过渡态及产物之间的相关性。能量计算采用双杂化泛函中的B2PLYP方法和def2-QZVPP基组,能量计算过程中考虑零点能矫正。本实验计算全部使用Gaussian09程序包完成[18],所研究的反应如下:
As+N2O→AsO+N2
(1)
As+NO2→AsO+NO
(2)
As+NO→AsO+N
(3)
1.2 动力学参数计算方法
经典过渡态的反应速率常数计算公式如下[19]:
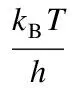
(4)
式中,kT为量子隧道修正系数;kB为Boltzmann常数,J/K;h为普朗克常数,J·s;QTS、QA、QB分别为过渡态TS、反应物A和反应物B的配分函数;Ea为反应活化能,kJ/mol;R为气体摩尔常数,J/(mol·K);T为热力学温度,K。其中,量子隧道修正系数计算公式如下:

(5)
式中,vm为反应路径振动频率,cm-1;c为光速,m/s。
2 结果与讨论
2.1 反应过程
采用B3LYP方法,在6-311G*基组水平上进行势能面扫描,然后得到势能面上的鞍点对应结构作为初猜,进行过渡态的寻找。然后,优化反应通道上所有驻点的几何构型,得到反应的微观进程,具体见图1-图3。

图 1 As+N2O→AsO+N2的反应过程示意图

图 2 As+NO2→AsO+NO的反应过程示意图
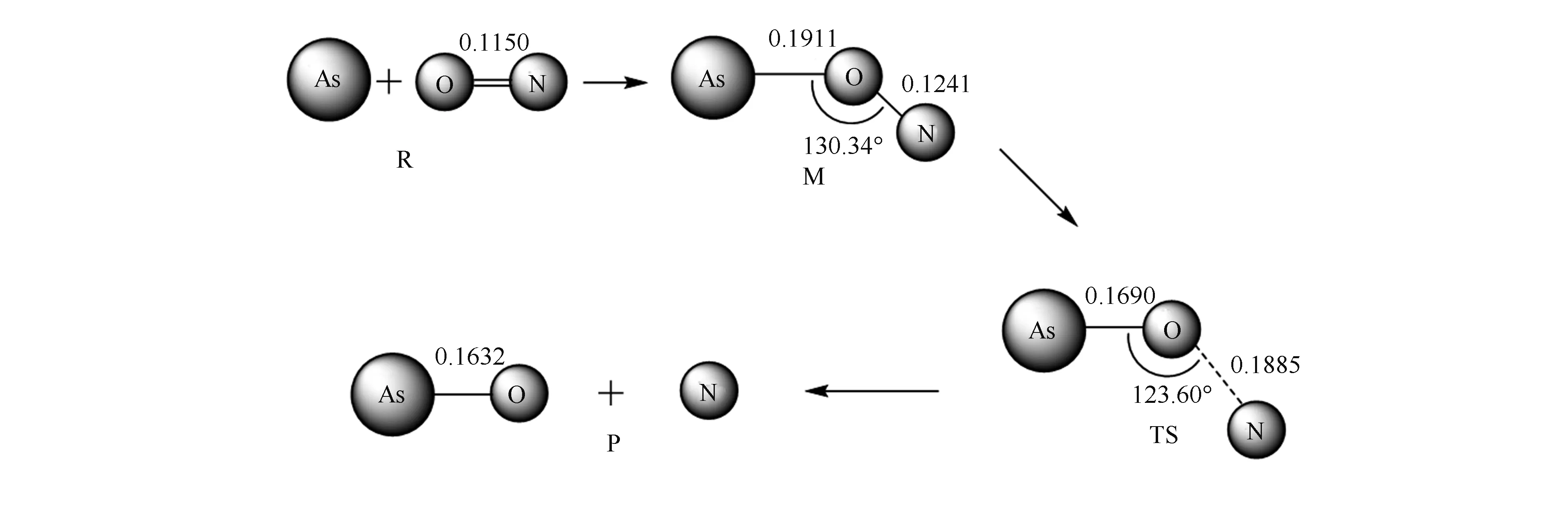
图 3 As+NO→AsO+N的反应过程示意图
由图1可知,反应式(1)的过程为:As+N2O→TS(AsONN)→AsO+N2,即As和O原子结合生成过渡态TS,然后形成产物AsO+N2,无中间体生成,只存在过渡态,是一步反应。在反应过程中,As-O键的长度逐渐减小(∞→0.2258 nm→0.1632 nm,∞表示距离超过了成键范围),O-N键的长度逐渐增加(0.1184 nm→0.1249 nm→∞),这说明As-O键的形成和O-N键的断裂是同时进行的。振动分析结果表明,过渡态有且只有一个虚频(-617.22 cm-1),在虚频振动模式下,O原子沿反应坐标向靠近As原子方向有显著的振动,说明该过渡态是准确的。
由图2可知,反应式(2)的过程为:As+NO2→M(AsONO)→TS(AsONO)→AsO+NO,即As和NO2的O原子结合生成稳定的中间体M,然后M经过渡态TS生成产物AsO和NO。在反应过程中,As-O键的长度逐渐减少(∞→0.1863 nm→0.1691 nm→0.1632 nm),O-N键的长度逐渐增加(0.1194 nm→0.1409 nm→0.1904 nm→∞),反映了As-O键的形成,而与之相邻的O-N键断裂的情况,分子结构的变化反映出了反应的微观过程。振动分析结果表明,中间体的振动频率全为正,是势能面上的稳定点;对过渡态进行振动分析发现有且只有一个虚频(-175.84 cm-1),且虚频的振动方向指向反应方向。
由图3可知,反应式(3)的过程为:As+NO→M(AsON)→TS(AsON)→AsO+N,在反应过程中,As-O键的长度逐渐减小(∞→0.1911 nm→0.1690 nm→0.1632 nm),表明As-O键逐渐形成;O-N键的长度逐渐增加(0.1150 nm→0.1241 nm→0.1885 nm→∞),表明O-N键逐渐断裂。振动分析结果表明,中间体的振动频率全为正,是势能面上稳定点;对过渡态进行振动分析发现有且只有一个虚频(-643.62 cm-1),说明过渡态是可信的,所研究的反应机理是合理的。
优化得到的反应物和生成物的分子结构见表1。由表1可知,本研究通过密度泛函中的B3LYP方法计算得到各分子稳定结构的参数与实验值吻合较好。
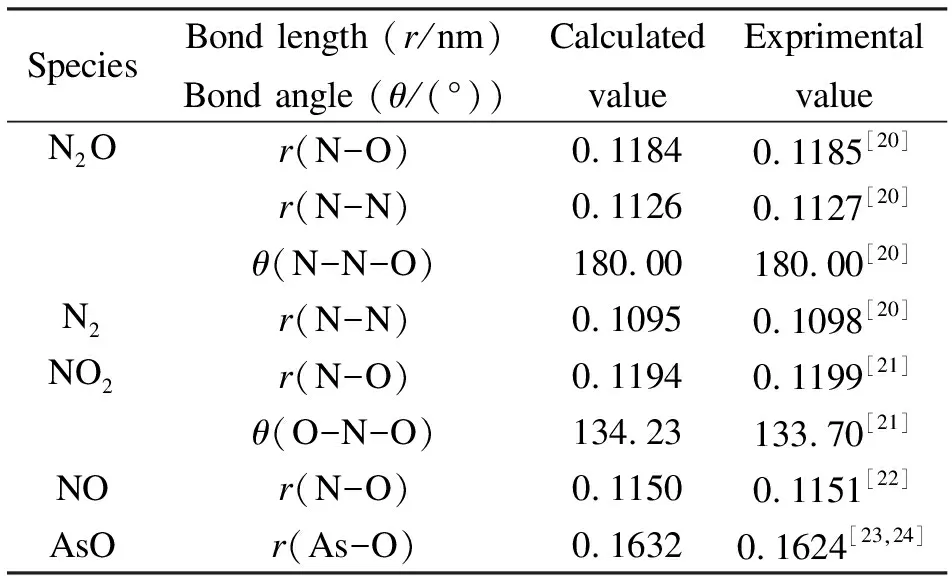
表 1 各反应物、生成物键长和键角的计算值及实验值
2.2 反应过程中的能量变化
反应物、中间体、过渡态和产物的能量、零点能、总能量Etot(B2PLYP/def2-QZVPP计算的能量加上零点能)和以反应物为参比的相对能量Erel见表2。
根据过渡态理论,活化能为过渡态与稳定的反应物(或中间体)之间的能量差,由表2可知,三种反应的活化能分别为78.45、2.58、155.85 kJ/mol,反应(2)的活化能最低,说明该反应容易进行。这是因为NO2有较强的氧化性[25],在反应过程中容易将砷单质氧化成AsO;然而,NO是一种较为稳定的气体,在反应过程中需要克服较大能量才能使N-O键断裂。在反应过程中,体系中的键长不断发生改变,即其体系的能量也不断改变。例如反应(1),当As原子靠近O原子时形成过渡态时,As-O键作用强烈,体系能量升高;过渡态与产物的As-O键长分别为0.2258和0.1632 nm,对应体系的能量分别为-2420.15112 (a.u.)和-2420.32382 (a.u.),可以看出As-O键长变化决定了体系的能量。
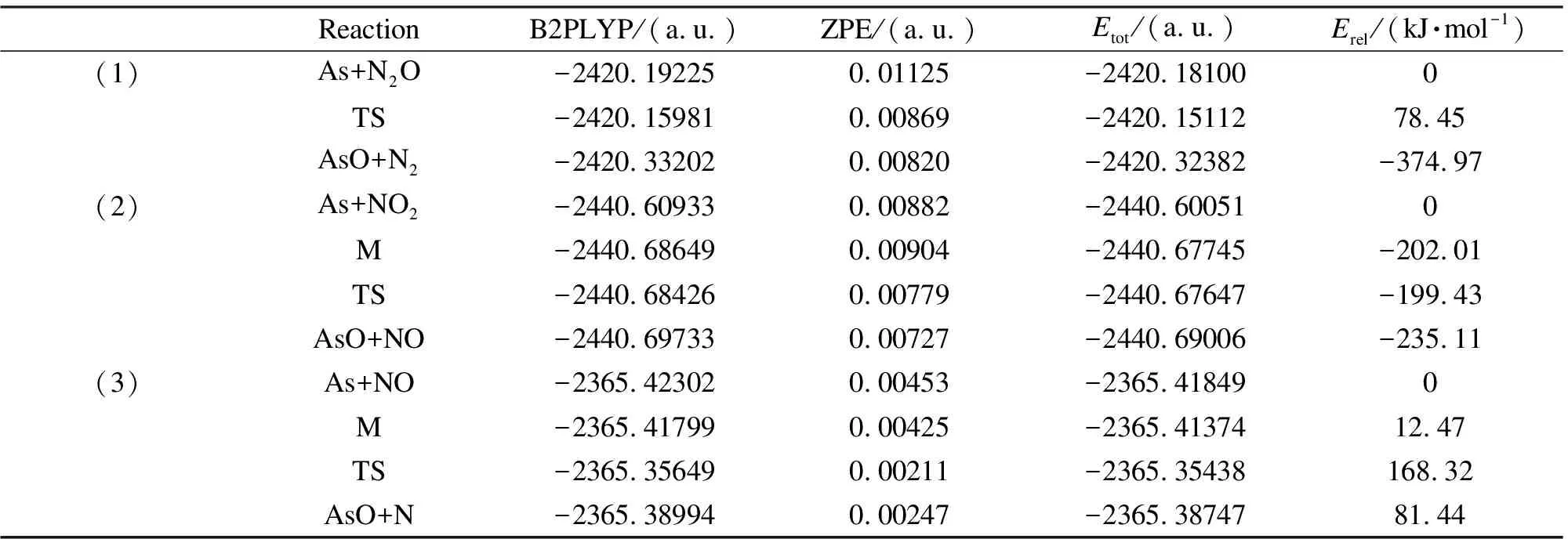
表 2 反应通道各驻点的能量
2.3 动力学分析
根据经典过渡态理论计算各反应在298、600、900、1200、1500、1800 K下的反应速率及指前因子,计算得到的结果见表3-表5。

表 3 不同温度As+N2O→AsO+N2的动力学参数值

表 4 不同温度As+NO2→AsO+NO的动力学参数值
由表3-表5可知,随着温度的升高,各反应的指前因子反应速率逐渐增大,但不同反应的增加幅度不一样。在考虑的温度范围内,反应(2)的速率始终大于1012,这说明该反应极快;反应(1)从1.15×10-3增加到7.26×108;反应(3)从4.95×10-15增加到9.33×108。

表 5 不同温度As+ NO→AsO+ N的动力学参数值
由k=A·exp(-Ea/RT)两边分别取对数,即lnk=lnA-(Ea/RT),以1000/T为横坐标,lnk为纵坐标将各反应的数据在同一坐标表现出来,具体见图4。由图4(a)可知,lnk与1000/T表现出良好的线性关系。图4(b)为各基元反应的lnk随温度的变化趋势。由图4(b)可知,各反应速率曲线随温度升高而增大,这与其他学者研究氮氧化物的还原是一致的[26]。反应(2)的反应速率曲线受温度的影响小,在考虑的温度范围内该反应瞬间完成。通过动力学参数可知,在所研究温度范围内,该反应的速率始终处于较高水平,即反应速率随温度的变化较小。另外,由于NO2活性高、有较强的氧化性[25],能与单质砷快速形成稳定的中间体,而中间体只需克服很小的能垒最后得到生成物,说明该反应极易发生。对于反应(1)和(3),在298-900 K,反应速率随温度的变化增加较快,当温度进一步升高,其增加的趋势有所减缓。
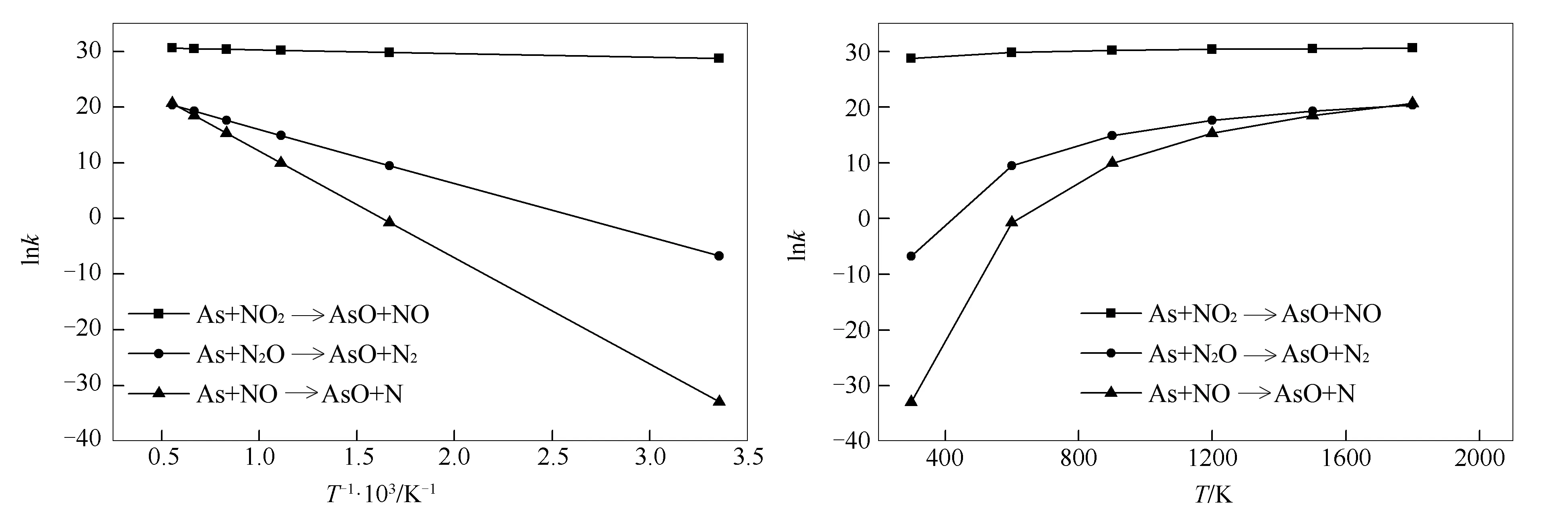
图 4 各反应lnk随反应温度的变化
通过线性拟合3种反应速率常数曲线得到其动力学参数,具体结果见表6。

表 6 反应动力学参数
3 结 论
As+NO2→AsO+NO反应的活化能为2.58 kJ/mol,说明反应极快;As+N2O→AsO+N2反应的活化能为78.45 kJ/mol,As+NO→AsO+N反应的活化能为155.85 kJ/mol,活化能较大,说明反应进行的相对较慢。
反应过程中,体系中的As-O键长主要决定了不同结构的能量,体系各原子间键长的变化反映出了反应的微观过程。
As与NO2的反应速率曲线受温度的影响小,在考虑的温度范围内该反应瞬间完成;而在298-900 K,砷与N2O和NO的反应,反应速率随温度的变化增加较快,当温度进一步升高,其增加的趋势有所减缓。
量子化学计算一种研究反应机理的有效手段。砷与氮氧化物的反应过程揭示了燃煤过程中砷的形态变化,为更经济有效地控制砷的排放提供了理论依据。
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29
汕头大学学报(自然科学版)(2020年4期)2020-12-14
山东工业技术(2018年9期)2018-05-26
中学化学(2017年5期)2017-07-07
中学化学(2016年4期)2016-05-30
股市动态分析(2015年12期)2015-09-10
中学化学(2014年1期)2014-04-23
汽车与新动力(2014年6期)2014-02-27
汽车与新动力(2012年4期)2012-03-25
汽车与新动力(2012年2期)2012-03-25
