
甲基丙二酸尿症合并亚甲基四氢叶酸还原酶缺陷1例报道并文献复习
2019-03-04 08:24梁瑞星郑宏牛冬鹤陆相朋
中国中西医结合儿科学 2019年1期
梁瑞星, 郑宏, 牛冬鹤, 陆相朋
甲基丙二酸尿症(methylmalonic aciduria,MMA)是一种常染色体隐性遗传病,是由甲基丙二酰辅酶A变位酶(Methylmalonyl-CoA mutase,MCM)缺陷或其辅酶腺苷钴胺素(AdoCbl)代谢障碍,导致机体代谢紊乱的一种有机酸尿症,包括单纯型MMA与MMA合并高同型半胱氨酸血症(合并型MMA)两种生化表型,我国MMA患者中约60%为合并型,其中cblC型国内MMA患者常见[1],为MMACHC基因突变导致。根据发病时间分为早发型和晚发型,其中晚发型较少见,多在1岁后发病,多以神经系统损害为主要表现,有些为单纯神经系统受累表现,缺乏多系统损害及代谢危象的特征性表现,易误诊,但经治疗后多数预后较好。亚甲基四氢叶酸还原酶(Methylenetetrahydrofolate reductase,MTHFR)缺陷是MTHFR基因突变导致血液叶酸、5-甲基四氢叶酸降低,同型半胱氨酸水平升高,蛋氨酸降低,引起一系列病理改变[2],是高同型半胱氨酸血症的常见类型之一,亦是一种罕见的常染色体隐性遗传性疾病。cblC型MMA和亚甲基四氢叶酸还原酶缺陷均为可治疗的遗传代谢病,国内外进行了较为深入的临床与实验室研究,共患两种遗传代谢病的临床表现较复杂,临床上容易漏诊误诊。本研究报道我国首例初诊疑诊急性播散性脑脊髓炎(acute disseminated encephalamyelitis,ADEM)的cblC型MMA共患亚甲基四氢叶酸缺陷的病例,并对其诊疗经过、基因特点进行分析。
1 临床资料
患儿,男,15岁,因“记忆力下降、行走功能障碍1年6月余”就诊。12岁半时感冒后出现体倦乏力、精神差,伴记忆力差,学习成绩下降,后逐渐出现双下肢无力,不能独站、独走,不伴意识障碍、感觉障碍及二便失禁。至省级医院查头颅及全脊髓MRI提示:(1)双侧大脑半球皮质FLATR信号略高(图1a);(2)左侧侧脑室体后部旁小片状稍长T1长T2信号(图1b);(3)T8~9水平脊髓内点片状T2压脂稍高信号(图1c),脑脊液检测未见异常,确诊为“急性播散性脑脊髓炎”,予规范治疗好转后出院。13岁半时再次出现行走困难,双下肢无力,行头颅核磁共振未见异常,当地间断予康复治疗,效果不明显,下肢肌张力进行性增高,遂转入河南中医院大学第一附属医院。父亲无临床表现,查同型半胱氨酸24.3 μmol/L,现口服叶酸、维生素B12治疗;母亲健康,非近亲结婚。患儿为第二胎第二产,足月顺产出生,围生期及新生儿期均无异常。生长发育同同龄儿童。体格检查:神志清,计算力、定向力无障碍。颅神经检查无异常。痉挛步态。双上肢肌力、肌张力正常。双下肢肌力Ⅳ级、肌张力(改良Ashworth分级)1+级,肌肉无明显萎缩及肥大。共济失调检查未见异常。深感觉、浅感觉未见异常。双侧膝腱、跟腱反射亢进,踝阵挛阳性,脑膜刺激征阴性,双侧巴氏征阳性。
实验室检查:(2015年12月外院)血常规、血生化、免疫球蛋白、补体均未见异常;脑脊液常规、生化、病原学及免疫性脑炎相关抗体检测未见异常。(2017年7月我院)同型半胱氨酸49.0 μmol/L(参考值0.0~15.0 μmol/L);血氨77 μmol/L(参考值9~30 μmol/L),乳酸2.75 mmol/L(参考值0.7~2.1 mmol/L);丙酮酸152.5 μmol/L(参考值20~100 μmol/L);叶酸11.04 μg/L(参考值3~17 μg/L);维生素B12>1 500 ng/L(参考值180~900 ng/L);尿有机酸分析提示尿甲基丙二酸29.63 mmol/mol肌酐(参考值0.2~3.6 mmol/mol肌酐)、3-羟基丙酸6.68 mmol/mol肌酐(参考值0.0~1.1 mmol/mol肌酐),血液游离肉碱16.17 μmol/L(参考值20.0~60.0 μmol/L),丙酰肉碱15.87 μmol/L(参考值1.00~5.00 μmol/L),C3/C0 0.15(参考值0.03~0.25);肌电图示四肢深感觉通路传导异常,双下肢视通路传导异常,双下肢神经源性肌损伤,考虑多发性运动、感觉神经损伤。疑诊MMA合并高同型半胱氨酸血症(遗传性?继发性?)。
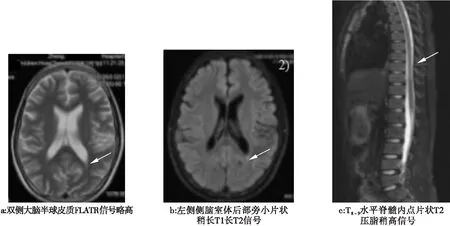
图1 患者头颅及全脊髓MRI结果
治疗与随访:患儿确诊后开始肌内注射羟钴铵每日1 mg,口服左卡尼汀每日2 g,甜菜碱每日3 g,叶酸片每日15 mg,正常饮食,同时继续家庭康复治疗。1个月后复查尿甲基丙二酸10.07 mmol/mol肌酐、3-羟基丙酸2.59 mmol/mol肌酐,血液游离肉碱88.26 μmol/L,丙酰肉碱15.87 μmol/L,C3/C0 0.18,同型半胱氨酸38.8 μmol/L。3个月随访,记忆力、运动功能及双下肢肌力明显改善,能独走、上下楼梯,双下肢肌张力仍较高,痉挛步态,无进行性加重。
基因分析:采取目标区域捕获+高通量测序验证代谢病相关基因分析(北京福佑龙惠遗传代谢病诊所)结果提示:患者在MMA合并同型半胱氨酸血症cblC型相关基因MMACHC外显子区域存在两处杂合突变点,分别为c.482G>A(p.R161Q)和c.626dupT(p.V209fs)。患者父母均为正常临床表型的携带者,其中c.482G>A突变来自其母;c.626dupT突变来自父亲。在亚甲基四氢叶酸还原酶缺乏型高胱氨酸尿症相关基因MTHFR外显子区域存在一处纯合突变c.665C>T(p.A222V)家系验证结果显示此纯合突变来自其父母,其父此位点为纯合突变,其母此位点为杂合突变。基因突变结果分析支持MMA合并高同型半胱氨酸血症及亚甲基四氢叶酸还原酶缺陷诊断。
2 讨论
cblC型是我国合并型MMA患者的最常见类型,由位于1p34.1上MMACHC基因突变所致的[2]。根据发病年龄又可分为早发型(发病年龄≤1岁)与晚发型(发病年龄>1岁)。晚发型与早发型相比,起病隐匿,多系统损害(包括脑、脊髓、周围神经、血液、肝、肾),运动障碍表现突出、智力损害相对较轻[3]。有时缺乏多系统损害或代谢危象的特征性表现,极易误诊。本例患者13岁6个月发病,发病前有感染的病史,发病初期有记忆力下降、精神差以及进行性运动障碍的神经系统症状,结合头颅核磁表现疑诊“ADEM”[4-6],经激素及丙种球蛋白等治疗病情有一过性好转[7],之后病情出现反复,复查头颅MRI提示轻度脑萎缩(见图2),脊髓MRI未见异常。本例患儿虽然发病初期有感染病史,早期有认知减退和轻微的、不典型的脑病症状;头颅、脊髓MRI提示大脑半球皮质、脑室体后部旁、脊髓内有小片状稍长T1长T2信号,但是不符合ADEM头颅MRI弥漫性、边界模糊、范围1~2 cm的病灶,主要累及皮质下脑白质的特点。1年后无诱因病情反复,但是复查核磁仅见轻度脑萎缩,未发现新的病灶,不支持ADEM诊断[8]。因此,我们疑诊遗传代谢病,进一步查血清同型半胱氨酸水平明显增高,尿中甲基丙二酸、3-羟基丙酸水平增高,血C0降低,C3明显增高、C3/C0正常,初步临床诊断MMA合并高同型半胱氨酸血症(遗传性?继发性?)。经基因检测发现基因MMACHC外显子区域存在c.482G>A和c.626dupT两处杂合突变点,确诊为合并型MMA。本例患者为晚发型MMA,cblC型MMA在我国晚发型MMA中最常见[1]。
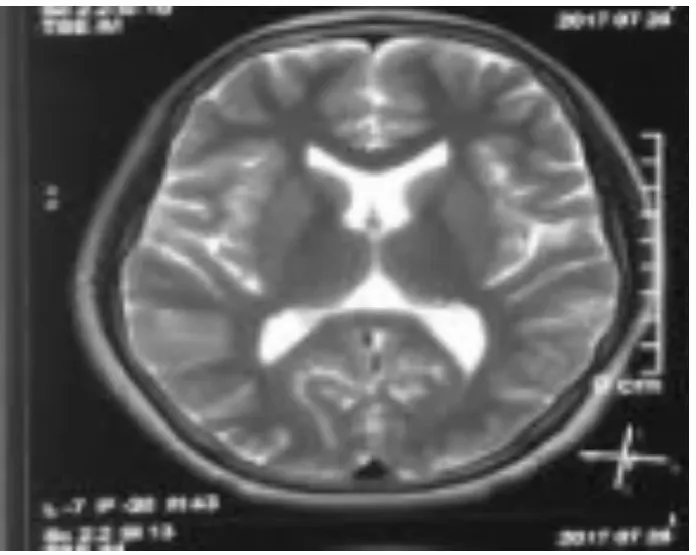
图2 患儿复查MRI示轻度脑萎缩
亚甲基四氢叶酸还原酶缺陷又称为高同型半胱氨酸血症Ⅱ型[9],是一种常染色体隐性遗传病。MTHFR是叶酸参与的同型半胱氨酸再甲基化过程中的关键酶。该酶缺陷导致5,10-亚甲基四氢叶酸转化为5-甲基四氢叶酸的转化受限,继而引起5-甲基四氢叶酸缺乏、高同型半胱氨酸血症和低蛋氨酸血症。多种多样的临床症状可以在新生儿期甚至在成人中出现。婴儿期表现最严重,包括肌张力减低、癫痫发作、昏迷。青少年或成人可能存在智力障碍、运动障碍和步态异常、癫痫发作、精神症状和血栓形成。其中脑血栓是晚发型患者较为常见的死亡原因[10],部分患者可终身无症状。本例患者起病隐匿,13岁6个月之前健康,发病后出现进行性下肢运动功能障碍。血同型半胱氨酸水平显著增高,符合高同型半胱氨酸血症,基因检测发现MTHFR基因665C>T纯合突变(已知突变),确诊为MTHFR缺陷。该患儿父亲无临床症状,同型半胱氨酸水平增高,基因检测同样为MTHFR基因665C>T纯合突变,属无症状性高同型半胱氨酸血症。
本例患者所患两种遗传性疾病cblC型MMA及高同型半胱氨酸Ⅱ型均可引起机体的同型半胱氨酸蓄积,而同型半胱氨酸是蛋氨酸的代谢产物,多种因素导致蛋氨酸代谢障碍引起体内同型半胱氨酸及其代谢物异常积累。在遗传因素中,多种酶缺陷可以导致同型半胱氨酸血症,均为常染色体隐性遗传病[11]。胱硫醚合成酶、亚甲基四氢叶酸还原酶、蛋氨酸合成酶三种酶参与其代谢,三者缺陷分别导致同型半胱氨酸血症Ⅰ、Ⅱ、Ⅲ型[12-13];在维生素B12代谢过程中,腺苷钴胺素和甲基钴胺素合成过程中需要cblC、cblD、cblE、cblF等蛋白参与[14],其中任何一种酶蛋白缺陷均可导致同型半胱氨酸血症。而不同病因的同型半胱氨酸血症应给予不同的饮食及药物治疗[15]。本例患者基因已证实同时存在MTHFR与cblC两种缺陷,血液蛋氨酸浓度正常。因此,应鼓励患者天然饮食,保证天然蛋白质的供给,同时补充羟钴铵、叶酸、甜菜碱、左卡尼汀。在疾病状态下由于代谢物蓄积,患者多存在厌食蛋白质等营养障碍,体内蛋氨酸水平会下降,严重时引起蛋氨酸缺乏,必要时补充蛋氨酸,多数患者预后良好[16-17]。
临床上共患两种隐性遗传病的发生非常少见。笔者通过查阅PUBMED文献及中国知网、万方数据库仅发现有5例MMA合并其他遗传代谢病的病例报告,其中1例患者诊断MMA 3年后发现先天性肾上腺皮质增多症[18];1例患者确诊为琥珀酰辅酶A合成酶缺乏继发出现的MMA[19];1例患者基因确诊为MMA合并肉碱增多症[20];另外2例均同时存在甲基丙二酸尿症cblC+cblD[21]与cblC+cblA[22]两种类型。甲基丙二酸尿症及MTHFR缺乏均为罕见隐性遗传病,本研究通过生化及基因分析确认了首例初诊为ADEM共患上述两种疾病的患儿。遗传代谢病的临床表现较为复杂,诊断困难,临床上容易漏诊误诊。正确的病因诊断是治疗的关键,早期诊断是改善预后的根本措施。
猜你喜欢
生物信息学(2022年3期)2022-11-12
中国现代医生(2022年21期)2022-08-22
中国饲料(2022年5期)2022-04-26
当代医药论丛(2021年24期)2022-01-20
西南农业学报(2021年10期)2021-12-14
医学美学美容(2021年21期)2021-11-20
猪业科学(2020年12期)2021-01-09
新疆大学学报(自然科学版)(中英文)(2020年2期)2020-07-25
医学新知(2019年4期)2020-01-02
中国医药科学(2017年9期)2017-08-04
