
应用双靶标CRISPR慢病毒载体在间充质干细胞中稳定敲除lncRNADANCR基因
2019-03-01 06:36钟小敏
中山大学学报(医学科学版) 2019年1期
彭 丹,钟小敏
(中山大学中山医学院干细胞与组织工程研究中心,广东广州510080)
长链非编码 RNA(long non-coding RNAs,lncRNAs)是一类碱基长度大于200 nt且不具备蛋白编码功能的RNA,在细胞的生长、分化、衰老、肿瘤的发生及转移中发挥着重要作用。LncRNA DANCR最初被报导在细胞分化过程中表达降低,且对维持细胞的未分化状态是不可缺少的[1]。随着对 DANCR(anti-differentiation noncoding RNA)功能研究的深入发现,DANCR也影响着肿瘤的发生与发展,如参与肿瘤干细胞的维持、促进肿瘤细胞侵袭与转移等[2]。近来有研究发现DANCR在间充质干细胞(mesenchymal stem cells,MSC)的成骨、成脂和成软骨的三向分化过程中也起到重要作用[3],DANCR的表达能促进MSC的成软骨分化而抑制其成骨分化[4-5],影响骨再生与软骨再生过程。CRISPR/Cas9[clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated 9(Cas9)]技术作为第三代基因编辑技术,与前两代基因编辑技术锌指核酸内切酶(zinc finger endonuclease,ZFN)和类转录激活因子效应物核酸酶(transcription activator-like effector nuclease,TALEN)相比,能对基因进行精准的编辑,且操作更为简便有效,近年来广泛应用于各种基因突变细胞株和基因突变动物的构建。lncRNA因不具备蛋白编码潜能,而以RNA分子的形式行使生物学功能,同时大部分lncRNA的活性作用位点尚未被研究清楚,所以可以预见,lncRNA分子上单一碱基的突变不一定能影响lncRNA整体结构和生物学功能,即应用CRISPR方法靶向单一位点难以阻断lncRNA功能。而常规的CRISPR/Cas9技术在进行两位点同时突变和小片段缺失时效率较低,常需要较大的工作量才能获得纯合克隆。本研究旨在通过双靶标CRISPR慢病毒载体,同时靶向于DANCR的5’及3’端,对DANCR进行全基因片段敲除,并构建慢病毒敲除载体,稳定敲除MSC中的DANCR基因。
1 材料与方法
1.1 细胞株及菌株
人胚肾上皮293FT细胞(购自Invitrogen公司)、骨髓来源间充质干细胞(本实验建系)及大肠杆菌菌株DH5α(购于天根生化科技有限公司)。
1.2 质粒及主要试剂
pX330A-1X2载体和pX330S-2载体(购于Addgene公司)、V6载体(由Professor Chunliang Li,St.Jude Children′s Research Hospital赠予)、慢病毒包装质粒pMDG、pRSV和pMDLg/pRRE(购于Addgene公司);DNA纯化回收试剂盒、质粒小提试剂盒和血液/细胞/组织基因组DNA提取试剂盒(购于天根生化科技有限公司);限制性核酸内切酶 BbsⅠ、KpnI和 EcoRI、T7 Endonuclease I(T7E1)及T4连接酶(购于New England BioLabs公司);Infusion kit(购自Takara公司);RNA提取试剂TRI-Reagent(购于MRC公司);逆转录试剂盒(购于赛默飞世尔科技公司);荧光定量PCR试剂SYBR(购于Roche公司)。
1.3 细胞培养
293FT细胞的培养:使用DMEM高糖培养基(Hyclone,美国),含10%胎牛血清(Hyclone,美国)及双抗(100 U/mL青霉素和0.1 mg/mL链霉素)(Hyclone,美国),置于37℃,体积分数5%CO2的细胞培养箱内培养;MSC的培养:使用DMEM低糖培养基(Hyclone,美国),含10%胎牛血清,及双抗(100 U/mL青霉素和 0.1 mg/mL链霉素),置于37℃,含体积分数5%CO2的细胞培养箱内培养。
1.4 单一sgRNA质粒构建
使用张锋实验室开发的在线设计sgRNA网站(http://tools.genome-engineering.org),设计靶向DANCR的sgRNA[6],挑选得分较高的两条靶向于5’端和两条靶向于 3’端的 sgRNA,5’端 1#:ACGCACAGCCAATCCCGGAT,5’端 2#:CTGGTT TGTGCGCCCGTCGC,3’端 1#:GTTAATTGACTA ACAACCCC,3’端 2#:GCTTCTCCACCAGTCGGA GG,合成4对寡聚核苷酸序列。如表1。
本研究中的序列合成及测序均交由生工生物工程股份有限公司完成,用BbsⅠ切割pX330A-1X2载体和pX330S-2载体,电泳后行切胶回收。将4对sgRNA分别退火并磷酸化处理后,5’端sgRNA连入线性化的pX330A-1X2载体(Amp抗性),3’端sgRNA连入线性化的pX330S-2载体(Kana抗性),连接产物转化到感受态细胞DH5α中,并使用相应抗性的LB平板筛选阳性克隆。挑取单克隆摇菌并用U6 promoter引物测序,测序正确的菌液转移至5 mL含相应抗性的液体LB培养基中摇菌过夜,取0.5 mL菌液与0.5 mL已灭菌的体积分数为30%甘油混合保存菌种,剩余菌液使用质粒小提试剂盒进行质粒提取。

表1 DANCR sgRNA寡核苷酸序列Table 1 Sequence of DANCR sgRNA oligonucleotide
1.5 质粒转染及sgRNA靶向性检测
取对数生长的293T细胞,在转染前一天接种至6孔板中,于含体积分数5%CO2的37℃培养箱于孵育 24 h,转染时将 2 μg 质粒加至 200 μL Opti-MEM中混匀,静置5 min后加入6 μL Megatran1.0,轻轻混匀并室温孵育15 min,均匀滴加至6孔板的一个孔中。24 h后更换新鲜培养基,转染后72 h使用基因组提取试剂盒进行基因组提取。根据sgRNA靶向位点设计靶向效率检测引物,DANCR 5’端检测引物-F:CAAGAGCACC AAAATCACCAACG ;DANCR 5’端检测引物-R:CGCAAAATTGTTACGGTGCCCAG ;DANCR 3’端检测引物-F:TGACCCTTACCCTGAATACTCTG CA ;DANCR 3’端检测引物-R:CACCAGGACAC CAAATCTCAAGAG。以转染后的293T细胞基因组DNA为模板进行普通PCR,得到的PCR产物跑胶,并使用DNA纯化回收试剂盒进行胶回收,取500 ng胶回收产物退火杂交,用T7E1进行酶切,酶切后电泳检测酶切条带。使用Image J软件分析各条带灰度值,根据公式 %Indels=[1-×100%计算打靶效率(其中a和b表示发生基因突变的DNA序列被切割后获得的短片段灰度值,c表示未发生基因突变而未被切割的长片段灰度值)。
1.6 将双靶标sgRNA连接至同一慢病毒载体
使用Golden gate assembly试剂盒将3’sgRNA连接至 5’sgRNA/pX330A-1x2载体:取 75 ng 5’sgRNA/pX330A-1x2与150 ng 3’sgRNA/pX330S-2混合,加入1 μL Golden mix和1 μL buffer,加双蒸水调整体积至20 μL,放入PCR仪,37 ℃ 5 min,16℃5 min,循环30次,将连接产物转化DH5α,使用Amp抗性的LB平板筛选阳性克隆,挑取克隆送至公司用测序引物1测序:CGGGCCATTTACCGT AAGTTATGTAACG。得到正确质粒后,设计引物将sgRNA及相关元件通过Infusion试剂盒克隆至V6载体(Amp抗性),PCR-F:GGTTAATTAAGGT ACCGCTGGCCTTTTGCTCACATGTG;PCR-R:CCT AGCTAGCGAATTCGTTATGTAACGGGTACCTCTA GGCC。以连入两条sgRNA序列的pX330A-1×2质粒为模板进行PCR,对PCR产物进行胶回收,用内切酶KpnI和EcoRI分别对PCR胶回产物和V6载体进行双酶切,酶切后电泳行切胶回收。取50 ng胶回收产物与100 ng线性化的V6载体,加入2 μL infusion 5 × mix,用水补齐体积至10 μL,50℃孵育1 h后取2 μL连接液进行转化,使用Amp抗性的LB平板筛选阳性克隆,挑取克隆送至公司用PCR-F测序。得到正确质粒后保存相应菌种并提取质粒用于下一步实验。
1.7 慢病毒包装及感染MSC
将慢病毒包装质粒:pMDG、pRSV和pMDLg/pRRE,按1∶1∶2比例混匀并调整浓度为1 μg/μL,取15.42 μL与8.58 μg构建好的双sgRNA/V6质粒混合,加至900 μL Opti-MEM中,室温静置5 min,然后加入72 μL转染试剂Megatran 1.0混匀,室温静置15 min,加入到T75中的293FT细胞(转染时细胞密度控制在80%~90%,转染前半换液),培养12 h后换液,分别收集换液后48 h和72 h的细胞培养液上清,上清900×g,4℃,离心15 min,用0.45 μm滤器过滤去除细胞碎片,取滤液50 000×g高速离心90 min沉淀病毒,去除上清后用1 mL无血清培养基重悬病毒沉淀,分装病毒保存于-80℃。MSC在感染病毒前一天铺板,感染时细胞密度为20%~30%,并加入polybrene使其终浓度为8 μg/mL,感染12 h后更换培养基,继续培养。
1.8 鉴定MSC中DANCR敲除效率
使用基因组提取试剂盒提取MSC基因组DNA,取等量基因组DNA作为模板,使用DANCR 5‘端检测引物-F和DANCR 3’端检测引物-R进行PCR,跑胶观察条带大小。使用Image J软件分析各条带灰度值,利用公式 %Indels=[1-×100%计算敲除效率(其中a表示被敲除后的小片段灰度值,b表示未发生敲除的野生型片段灰度值)。将条带进行切胶回收,直接将胶回收产物用PCR-F进行测序,并与DANCR基因参考序列进行比对。
1.9 RNA提取、逆转录及实时荧光定量PCR检测MSC中DANCRmRNA表达水平
RNA提取前将细胞接种至6孔板中,待细胞密度达到80%~90%,移除培养基并用分别加入1 mL预冷PBS清洗细胞,移除PBS后每孔加入1 mL TRI-Reagent裂解细胞,室温静置10 min后将细胞吹落,转移至1.5 mL离心管中。每管加入200 μL三氯甲烷并颠倒6~8次充分混匀,于冰上静置3 min,此时可见液体分层,11 000×g,4℃,离心15 min,离心结束后液体分为三层,其中上层透明液体中含RNA。小心吸取400 μL上层透明液体至不含RNA酶的1.5 mL离心管中,加入等体积异丙醇并颠倒6~8次充分混匀,于冰上静置10 min后,11 000×g,4℃,离心15 min。离心后可在管底见到白色RNA沉淀,弃上清,每管加入1 mL体积分数为75%乙醇漂洗沉淀,11 000×g,4℃,离心5 min后弃上清,晾干RNA沉淀至边缘透明,视沉淀多少加入20~30 μL不含RNA酶的去离子水溶解沉淀。测定浓度,取1 μg RNA使用逆转录试剂盒按说明书进行逆转录。逆转录后得到cDNA后按1∶1稀释后进行实时荧光定量PCR,设计实时荧光定量PCR引物,DANCR-F:GCCACTATGTAGCGGGTTTC;DANCR-R:GCCT GTAGTTGTCA ACCTGC,荧光定量PCR每孔体系为:cDNA 1 μL、SYBR 5 μL、引物 2 μL、水2 μL,总体系10 μL,荧光定量PCR仪检测,数据分析。
1.1 0 统计学方法
采用SPSS 17.0进行单因素方差分析,P值显著性水准均为0.05(双侧检定),P<0.05表示差异有统计学意义。
2 结果
2.1 单一sgRNA载体构建及打靶效率
利用麻省理工学院张锋实验室开发的在线sgRNA工具设计网站,靶向DANCR的5’和3’端各设计两条sgRNA,并分别命名为sgRNA5’-1、sgRNA5’-2 和 sgRNA3’-1、sgRNA3’-2。 各sgRNA的靶向序列在DANCR基因中的位置如图1A所示。通过退火使寡核苷酸单链形成双链,然后将退火产物分别连入pX330A-1X2载体和pX330S-2载体,使用U6 promoter引物测序,测序正确质粒的测序结果如图1B-1E所示。扩增正确质粒瞬时转染293FT,提取293FT基因组DNA,经T7E1酶切后检测打靶效率,结果如图1F所示,sgRNA5’-1、sgRNA5’-2 和 sgRNA3’-1、sgRNA3’-2的打靶效率分别为6.62%、4.96%、5.60%、5.60%。
2.2 双靶标载体构建
双sgRNA载体构建示意图如图2A所示,通过Golden gate assembly试剂盒将 sgRNA5’-1和sgRNA3’-1连至同一个载体、sgRNA5’-2和sgRNA3’-2连接至同一个载体,使用测序引物1进行测序,扩增测序正确的质粒,PCR扩增双sgRNA相关元件,并连接至慢病毒CRISPR载体V6,分别命名为1#DANCR KO/V6和2#DANCR KO/V6,菌液PCR鉴定阳性克隆,选取阳性克隆提取质粒并使用PCR-F进行测序,结果如图2B、2C所示。
2.3 检测双靶标慢病毒载体在MSC中对DANCR的敲除效率
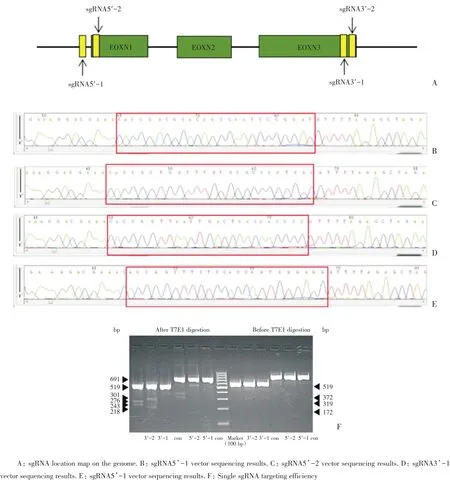
图1 单一sgRNA载体构建及打靶效率Fig.1 Single sgRNA vector construction and targeting efficiency
在293FT中进行慢病毒包装,V6载体带有mcherry报告基因(如图2A所示),故可通过观察荧光强度判断转染效率。293FT荧光与明场合成图如图3A所示,收集病毒液并浓缩后感染MSC,感染后的MSC荧光与明场合成图如图3B所示,细胞感染阳性率较高,故未进行流式分选。提取MSC总RNA检测DANCR mRNA表达水平变化,1#载体敲除后MSC的DANCR表达水平下降至原来的(0.20±0.05)倍,2#载体敲除后MSC的DANCR表达水平下降至原来的(0.13±0.01)倍,结果如图3C所示(样本量为3;1#载体敲除后MSC的DANCR表达水平与对照组相比较,F=623.92,P=0.002;2#载体敲除后MSC的DANCR表达水平与对照组相比较,F=21053,P<0.001)。提取MSC基因组DNA进行DANCR敲除检测,结果如图3D所示。用Image J分析DANCR敲除效率,1#和2#载体的敲除效率分别为(58.4±11.0)%和(81.0±6.2)%,结果如图3E所示(样本量为3;1#载体敲除效率与对照组相比较,F=7 299.1,P<0.001;2#载体敲除效率与对照组相比较,F=51 216,P<0.001)。图3D中敲除后的小片段PCR产物经过回收纯化,将产物直接使用PCR-F引物进行测序,并与DANCR基因参考序列进行比对,结果显示MSC DANCR KO 1#细胞DANCR缺失片段为1 743 bp,MSC DANCR KO 2#细胞DANCR缺失片段为1 601 bp,且缺失片段为5’sgRNA靶点与3’sgRNA靶点之间序列,比对结果如图3F-3G所示。
3 讨论
CRISPR/Cas9技术是基于细菌等原核生物利用CRISPR和Cas蛋白特异降解外源入侵核酸这一原理建立的基因编辑技术[7]。与TALEN和锌指等基因编辑技术相比,CRISPR/Cas9技术具有操作简单快速、成本低、效率高、作用位点更广的优点,自该技术建立以来便成为生命科学领域常用的研究工具,广泛应用于各种基因突变细胞株和基因突变动物的构建[8]。
长链非编码RNA是一类碱基长度大于200 nt且不具备蛋白编码功能的RNA,人类基因组中已有超过15 000个lncRNA被测序发现,且GENCODE中的注释也在不断更新。在很长的一段时间里lncRNA都被认为是转录产生的垃圾RNA,但近来的研究表明lncRNA在调节核染色质结构和基因表达中发挥重要作用,并参与细胞的生长分化、疾病的发生等生理及病理过程。lncRNA-DANCR最初是在钙离子诱导人初级角质细胞分化的过程中被发现,通过RNA测序结果发现在这一分化过程中DANCR的表达下调[1]。DANCR位于4q12染色体上,由三个外显子组成,长度为915 bp。近年来的研究表明DANCR与肿瘤的发生与进展有着密切联系:通过与抑制CTNNB1的microRNA结合,解除microRNA对CTNNB1的抑制作用以增加肝癌细胞的干性,促进肝癌的进展[9];且DANCR受原癌基因MYC的调控,在多种肿瘤细胞中表达上调,抑制细胞周期抑制因子P21的表达[10];DANCR的表达介导肿瘤细胞的耐药性且与患者的不良预后相关[11]。相对于DANCR在肿瘤中的功能研究,DANCR在干细胞分化过程中的具体功能研究较少。已有研究表明下调牙髓干细胞中的DANCR表达促进其成骨、成脂和神经分化[12],且DANCR参与MSC的三向分化过程,能促进MSC的成软骨分化[13-14]、抑制其成骨分化,而MSC经历骨祖细胞、前成骨细胞最终分化为成骨细胞的过程对骨折损伤和一些疾病后的骨再生过程有着重要意义[15]。
本研究通过分别靶向DANCR的5’和3’末端位点设计sgRNA,并将两条sgRNA克隆进同一慢病毒CRISPR载体,能达到较高的敲除效率。普通CRISPR技术常需要挑取较多克隆才能得到完全敲除的目的克隆,常带来较大的工作量,并耗费大量的培养基。我们构建的双靶标慢病毒敲除载体能极大地提高敲除效率,分析原因:将两条sgRNA连接在同一载体上,能保证两套CRISPR系统能在同一细胞中表达,且通过慢病毒载体能整合至细胞的基因组中,保证敲除效率。已有的关于DANCR在MSC中的功能研究均使用干扰RNA敲低DANCR的表达,本研究首次构建了DANCR敲除的MSC细胞系。稳定敲除DANCR的MSC与使用干扰RNA敲低DANCR基因表达的MSC相比,干扰RNA只能降低DANCR的表达而无法完全清除,通过CRISPR/Cas9技术获得DANCR基因敲除的MSC细胞株,完全缺失DANCR基因功能,有利于更深入、准确的研究DANCR在MSC中的功能。DANCR敲除载体的构建为进一步深入研究DANCR在不同细胞中的功能奠定了基础,也可用于DANCR敲除动物模型的构建。
综上所述,本研究所应用的双靶标sgRNA的CRISPR敲除慢病毒载体设计方案为长片段的敲除提供了新的思路,可用于构建其它lncRNA的CRISPR敲除载体。本研究建立的DANCR敲除的MSC细胞系,可用于深入研究DANCR在MSC中的功能。
猜你喜欢
环球时报(2022-09-20)2022-09-20
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
中老年保健(2022年1期)2022-08-17
保健医苑(2022年5期)2022-06-10
江西农业学报(2021年4期)2021-04-20
昆明医科大学学报(2021年2期)2021-03-29
今日农业(2020年24期)2020-12-15
三农资讯半月报(2020年11期)2020-06-21
小资CHIC!ELEGANCE(2015年14期)2015-09-23
