
BMN-673类似物的合成
2019-02-20 08:52杨梦然冯权武尹传奇
武汉工程大学学报 2019年1期
杨梦然,胡 旭,冯权武,吴 莉,傅 晶,尹传奇*
1.武汉工程大学化学与环境学院,湖北 武汉 430205;
2.湖北惠生药业有限公司,湖北 咸宁 437000
PARP(poly ADP-ribose polymerase)是一种聚腺苷二磷酸核糖聚合酶,参与染色体重塑、调控细胞凋亡和分裂等过程,被称为“DNA的守护神”[1-2]。在正常细胞中,受损的DNA修复是一个重要的过程,但在肿瘤细胞中,DNA损伤显著,PARP呈过表达状态,催化活性增强,通过抑制PARP的活性来切断DNA损伤修复通路,可达到杀死肿瘤细胞的目的[3-4]。所以,PARP抑制剂已成为抗癌药物研究的热点之一。
目前已进入临床的PARP抑制剂有5种类型[5],其中talazoparib类由于拥有较大的结构和立体特异性,导致它对PARP-1和PARP-2的捕获性最高。Biomarin公司研发的BMN-673(图1)是一种新型的口服PARP抑制剂,其口服计量小,有良好的生物利用度,是已报道最有效的talazoparib[6-7]。其作为可注射纳米制剂正在进一步研究[8],在治疗BRCA突变、HER2阴性、转移性晚期乳腺癌试验正处于 III期临床阶段[9-10],在治疗胃癌[11]、高级浆液性卵巢癌[12]、子宫内膜癌[13-14]、骨髓增生性肿瘤[15]、携带有害BRCA突变的实体瘤[16]的试验已进入临床研究阶段,与放射疗法结合用于治疗各种癌症的研究也正在试验中[17]。
在 BMN-673分子结构中,1-甲基-1H-1,2,4-三唑-5-基可提高药物的水溶性和细胞活性,对氟苯基为富电子的芳香环,与Try-907发生π-π作用增强抑制活性,芳环上的两个氟原子可增强酶活性和透膜性,该化合物在酶水平上的活性和细胞活性均得到了提高[18-19]。本研究以BMN-673结构为基础,结合PARP抑制剂的构效关系设计合成了9个BMN-673类似物(见图2),以期筛选出有生物活性的化合物。在这些类似物中,R2基团均为富电子芳香环,除4a1外,其它分子中R2基团都连有含氧或含氮的杂环以增强分子水溶性;4c1和4c2中R1为苯基,4b1和4b2中R1为对氟苯基,可对比考察氟原子对分子生物活性的影响;4d中R1基团可以增强水溶性和口服生物利用度[20]。
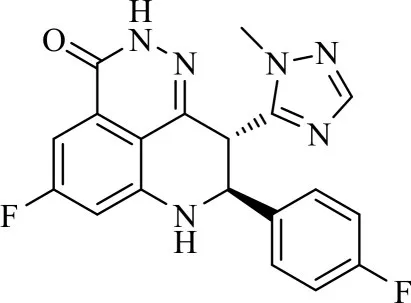
图1 BMN-673分子结构Fig.1 Molecular structure of BMN-673
BMN-673的合成路线有以下两种:(1)以4-氨基-6-氟异苯并呋喃-1(3H)-酮为原料,经亲核加成缩合、开环、合环三步反应生成目的产物。该合成路线中第一步反应需在无水条件下进行,并且反应生成的水需要用无水硫酸镁除去,耗时长。而后两步反应的收率分别为1.6%和29%,造成总收率仅为 0.3%[21-22];(2)以 6-氟-4-硝基-3H-异苯并呋喃-1-酮为原料,经四步反应合成[23](图 2),反应总收率为6.4%~61%。本研究采用第二种方法合成BMN-673类似物。
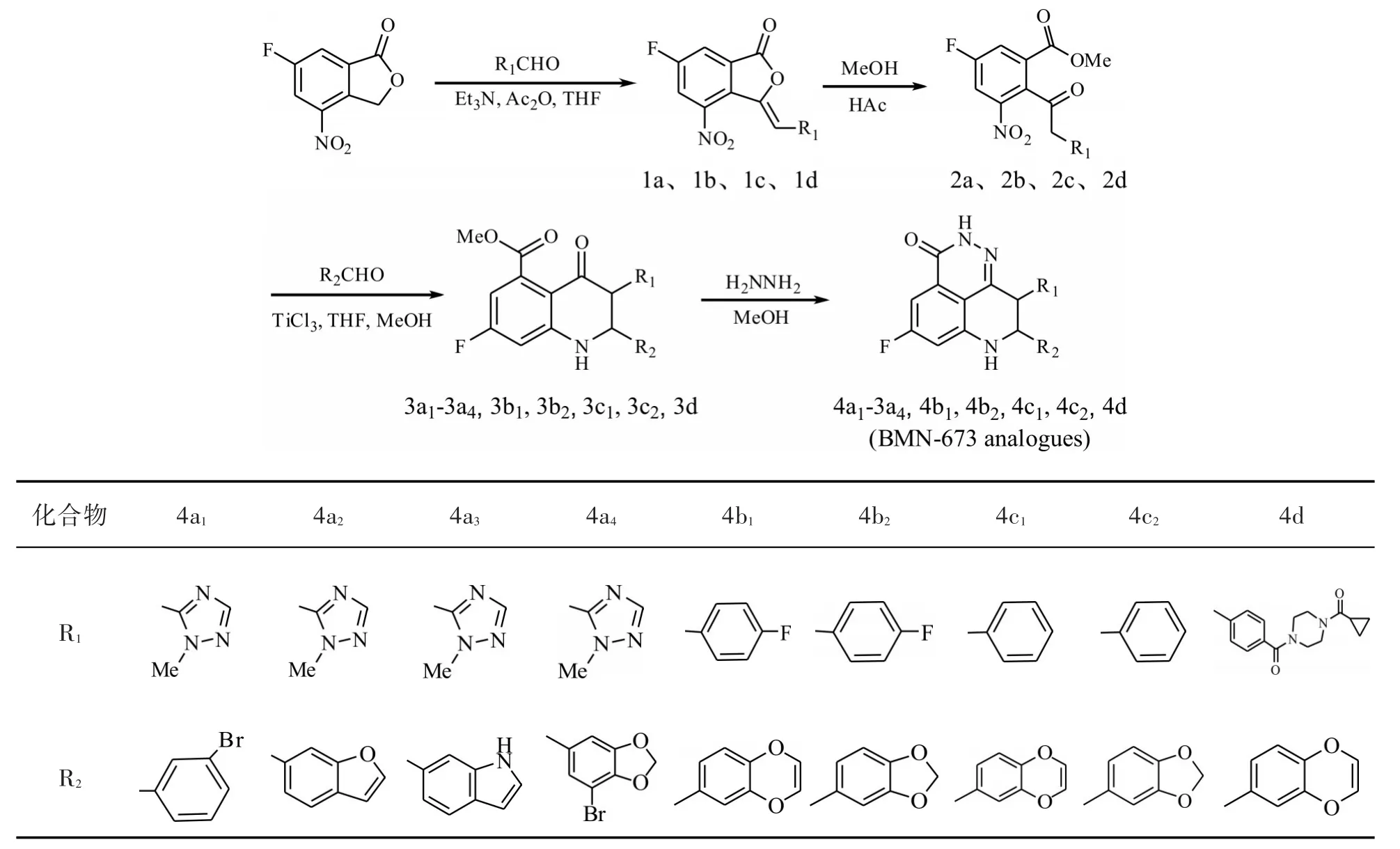
图2 BMN-673类似物的合成路线Fig.2 Synthetic route of BMN-673 analogues
1 实验部分
1.1 试剂与仪器
DPX 300核磁共振仪(德国Bruker公司);Tracems 2000色谱-质谱联用仪(美国Finnigan公司);EL III型元素分析仪(德国Vario公司);RY-1熔点检测仪(温度计未经校正)(天津天光光学仪器有限公司);LC-20 AT型高效液相色谱仪(HPLC,日本岛津公司)。
所用试剂均为国产市售分析纯。
1.2 实验过程
以 8-(3-溴苯基)-5-氟-9-(1-甲基-1H-1,2,4-三唑-5-基)-2,7,8,9-四氢-3H-吡啶并[4,3,2-de]-酞嗪-3-酮(4a1)的合成为例进行介绍。
1.2.1 (Z)-6-氟-3-((1-甲基-1H-1,2,4-三唑-5-基)亚甲基)-4-硝基异苯并呋喃-1(3H)-酮(1a)的合成
在 25 mL 三颈瓶中,将 0.50 g(2.5 mmol)6-氟-4-硝基-3H-异苯并呋喃-1-酮溶于 5 mL的 1,4-二氧六环中,依次加入1 mL(7.3 mmol)三乙胺、2 mL(21.2 mmol)乙酸酐和 0.55 g(5.0 mmol)1-甲基-1H-1,2,4-三唑-5-甲醛,搅拌,控制温度 105 ℃反应4 h,反应液由浅黄色变为深红棕色。浓缩80%的溶剂,冷却残留液,过滤,固体用甲基叔丁基醚淋洗,得到0.66 g黄色固体,产率为90%;mp:271~274 ℃ ;MS(m/z):291.05[M+H]+;1HNMR(400 MHz,CDCl3)δ 7.68(m,1H),7.45(m,1H),6.87(m,1H),6.69(s,1H),3.47(s,3H);13CNMR(100 MHz,CDCl3)δ165.3,161.1,147.8,145.3,143.1,141.2,125.9,124.1,120.1,115.9,96.8,33.9。 EA(C12H7FN4O4),Calcd:C 49.66,H 2.43,N 19.31;Found:C 49.70,H 2.42,N 19.29。
1.2.2 5-氟-2-(2-(1-甲基-1H-1,2,4-三唑-5-基)乙酰基)-3-硝基苯甲酸甲酯(2a)的合成 将0.66 g(2.3 mmol)化合物 1a、0.2 mL(3.5 mmol)醋酸和2 mL甲醇加入至25 mL三颈瓶中,搅拌,升温至50~60℃反应10 h,溶液由黄色浑浊状变为黄色透明,将反应液浓缩,然后柱层析,得到0.63 g黄色固体,产率为 86%;mp:280~283 ℃;MS(m/z):323.07[M+H]+;1HNMR(400 MHz,CDCl3)δ8.20(d,1H),8.01(d,1H),7.83(s,1H),4.84(s,3H),4.03(s,2H),4.00(s,3H);13CNMR(100 MHz,CDCl3)δ 198.7,166.2,165.3,163.0,150.1,145.3,129.4,125.1,120.1,110.1,51.2,35.5,35.2;EA(C13H11FN4O5),Calcd:C 48.45,H 3.44,N 17.39;Found:C 48.50,H 3.43,N 17.42。
1.2.3 2-(3-溴苯基)-7-氟-3-(1-甲基-1H-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-甲酸甲酯(3a1)的合成 将0.60 g(1.9 mmol)化合物2a、0.74 g(4.0 mmol)间溴苯甲醛、6 mL四氢呋喃和1 mL甲醇加入至50 mL三颈瓶中,搅拌下缓慢滴加12 mL三氯化钛盐酸溶液(质量分数20%,三氯化钛溶于2 mol/L盐酸),反应液呈黑色透明,室温搅拌,溶液由黑色透明变为深黄色,反应6 h后,加20 mL水淬灭,混合液用乙酸乙酯萃取,有机相依次用饱和碳酸氢钠和饱和亚硫酸氢钠洗涤,无水Na2SO4干燥,浓缩得 0.70 g黄色油状物,产率 82%;MS(m/z):459.27[M+H]+;1HNMR(400 MHz,CDCl3)δ8.04(s,1H),7.44-7.42(m,2H),7.29-7.23(m,2H),6.84(d,1H),6.69(d,1H),5.31(s,1H),5.13(d,1H),4.13(d,1H),3.83(s,3H),3.71(s,3H);13CNMR(100 MHz,CDCl3)δ190.1,166.9,165.3,160.1,150.2,148.2,140.2,130.2,128.7,121.3,120.9,120.1,118.3,116.0,103.4,104.1,58.7,52.3, 50.5, 32.4;EA (C20H16BrFN4O3),Calcd:C52.30,H 3.51,N 12.20;Found:C 52.09,H 3.50,N 12.15。
1.2.4 8-(3-溴苯基)-5-氟-9-(1-甲基-1H-1,2,4-三唑-5-基)-2,7,8,9-四氢-3H-吡啶并[4,3,2-de]-酞嗪-3-酮(4a1)的合成 将 0.70 g(1.5 mmol)化合物3a1、6 mL甲醇和1.5 mL水合肼加入至25 mL三颈瓶中,室温搅拌过夜,有固体析出,抽滤,甲醇洗涤,得到0.50 g浅黄色固体,产率为75%(HPLC含量99.2%)。mp:321.4~325.0 ℃;MS(m/z):441.26[M+H]+;1HNMR(400 MHz,DMSO-d6)δ12.38(s,1H),7.96-7.61(m,4H),7.88(d,1H),7.28(s,1H),7.08(d,1H),5.37(s,1H),5.04(d,1H),4.80(d,1H),3.69(s,3H);13CNMR (100MHz,DMSO-d6)δ 165.3,161.9,155.1,153.1,150.3,148.7,144.7,132.8,129.1,128.9,128.1,127.3,125.6,109.1,102.9,100.8,61.2,44.3,35.8;EA(C19H14BrFN6O),Calcd.:C51.72,H 3.20,N 19.05;Found:C 51.92,H 3.19,N 19.13。
2 结果与讨论
2.1 化合物1a-1d的合成
在碱性条件下,6-氟-4-硝基-3H-异苯并呋喃-1-酮中苄位上的氢原子被拔去,形成碳负离子,该碳负离子由于苯环上氟原子和硝基的吸电子效应而稳定。碳负离子与1-甲基-1H-1,2,4-三唑-5-甲醛发生亲核加成反应,然后脱去一分子水生成化合物1a,乙酸酐起吸水作用。
文献[23]报道,上述反应采用四氢呋喃做溶剂,回流反应2 h,产率为78%,但实验时发现此反应实际需要10 h才能完成。因此,本研究探索了几种溶剂对反应的影响(见表1)。当溶剂为丙酮时,基本无反应;当溶剂为甲醇时,反应24 h产率仅为10%。当溶剂为1,4-二氧六环时,反应4 h,产率即可达90%。因此,本反应选用1,4-二氧六环做溶剂。反应完成后,浓缩质量分数80%的1,4-二氧六环,冷却后有固体析出。在洗涤滤饼时,若用乙酸乙酯做淋洗剂[23],由于乙酸乙酯对产物1a有一定的溶解性,会影响到产物的收率。实验中改用溶解性低的甲基叔丁基醚洗涤,既可避免产品的损失,又由于甲基叔丁基醚沸点较低,产品易干燥、纯化简便。化合物1b-1d用类似的方法合成,相关表征数据见表2。
由于4-溴甲基苯甲醛、对羟甲基苯甲醛和4-吗啉甲基苯甲醛中均含有苯环,与Ty896和Ty907形成π-π相互作用,可增强体外活性,其中4-溴甲基苯甲醛可以增加细胞活性;对羟甲基苯甲醛可以增加细胞活性和水溶性;4-吗啉甲基苯甲醛含有两个氮原子可以提高水溶性[20],所以研究中考虑将这三种醛与6-氟-4-硝基-3H-异苯并呋喃-1-酮发生亲核加成反应。但遗憾的是反应没有发生,原因是这三个醛中苯环对位基团的给电子效应降低了羰基碳的亲电性。

表1 不同反应条件对合成化合物1a的影响Tab.1 Effects of reaction conditions on formation of compound1a
2.2 化合物2a-2d的合成
在酸性催化下,1a分子中的五元内酯与甲醇发生酯交换开环反应,并异构化为酮酯2a。文献[23]报道,采用醋酸和甲醇开环反应4 h,产率达到99%,但实验时发现需要反应10 h且产率最高达到86%。实验中探讨了乙酸、盐酸和三氟乙酸对酯交换开环反应的影响,如表3所示,醋酸的催化效果最好。用类似的方法合成2b-2d,相关表征数据见表4。

表2 化合物1b-1d的相关表征数据Tab.2 Characterization data of compounds 1b-1d

表3 几种酸对合成化合物2a的影响Tab.3 Effects of several acids on formation of compound 2a
由于1-甲基-1H-咪唑-2-基具有较好的亲水性,拟利用上述方法合成R1为1-甲基-1H-咪唑-2-基的类似物,但实验中发现该类分子中的内酯难以开环。原因在于,整个分子为共轭体系,化合物1a-1d分子中 R1基团 1-甲基-1H-1,2,4-三唑-5-基、对氟苯基、苯基和4-(4-(环丙烷甲酰基)哌嗪-1-甲酰基)苯基具有吸电子效应,降低了五元内酯上羰基碳的电子云密度,易于亲核试剂(甲醇)的进攻;而1-甲基-1H-咪唑-2-基具有给电子效应,增加了五元内酯羰基碳的电子云密度,使得亲核取代反应难以进行。
2.3 化合物 3a1-3a4、3b1、3b2、3c1、3c2和 3d的合成
利用TiCl3将2a分子中的硝基还原为氨基,生成物与3-溴苯甲醛发生羟醛缩合反应,生成的羟基与氨基脱水生成化合物3a1。用类似方法合成3a2、3a4、3b1、3b2、3c1、3c2和 3d。但在合成 3a3时,需先将1H-吲哚-6-甲醛分子上的-NH基团通过酰基化保护起来[25]。相关表征数据见表5。
2.4 化合物 4a1-4a4、4b1、4b2、4c1、4c2和 4d的合成
在甲醇溶液中,水合肼同时与3a1中的酯基和酮羰基作用,得到关环产物4a1。用类似的方法合成 4a2-4a4、4b1、4b2、4c1、4c2和 4d,相关表征数据见表6。

表4 化合物2b-2d的相关表征数据Tab.4 Characterization data of compounds 2b-2d

表 5 化合物 3a2-3a4、3b1、3b2、3c1、3c2和 3d 的相关表征数据Tab.5 Characterization data of compounds 3a2-3a4,3b1,3b2,3c1,3c2and 3d

续表5

表 6 化合物 4a2-4a4、4b1、4b2、4c1、4c2和 4d 的相关表征数据Tab.6 Characterization data of compounds 4a2-4a4,4b1,4b2,4c1,4c2and 4d

续表6
3 结语
本研究以6-氟-4-硝基-3H-异苯并呋喃-1-酮为原料经亲核加成反应、内酯酯交换开环、羟醛缩合和酰肼环化四步反应合成了9个新的BMN-673类似物,总产率为33%~48%。在第一步亲核加成反应中,以1,4-二氧六环为溶剂,不仅缩短了反应时间,也提高了转化率;后处理过程经浓缩、冷却析晶,固体用甲基叔丁基醚洗涤,既可减少产品损失,又有利于产品干燥和纯化。在第二步内酯酯交换开环反应中,醋酸为最佳催化剂,内酯分子中R1基团的吸电子能力越强,五元内酯越易开环。所合成的9个BMN-673类似物的生物活性有待进一步研究。
猜你喜欢
现代食品(2022年11期)2022-12-06
现代食品科技(2022年5期)2022-05-30
天津建设科技(2022年2期)2022-05-05
动物营养学报(2022年1期)2022-02-20
今日农业(2021年2期)2021-11-27
农药学学报(2021年4期)2021-08-26
今日农业(2020年23期)2020-12-31
山西农业科学(2020年11期)2020-11-19
火工品(2019年3期)2019-09-02
北京理工大学学报(2016年6期)2016-11-22
