
聚合物带隙及吸收光谱计算精度的研究
2019-02-19 01:29,,,,,,
郑州大学学报(理学版) 2019年1期
, , , , , ,
(1.郑州大学 物理工程学院 河南 郑州 450001; 2.郑州大学 现代分析技术与计算中心 河南 郑州 450052)
0 引言
近年来,本体异质结聚合物太阳能电池的研究得到快速发展,目前电池的光电转换效率已超过10%[1-2].理想的本体异质结电池活性层给体材料应满足吸收光谱与太阳辐射谱有较高的匹配度、高的载流子迁移率、给受体间好的能级匹配和制作过程中有较好的溶解以及成膜性[3-9].本体异质结太阳能电池活性层通常由电子给体和电子受体共混制备而成.电子受体材料一般是富勒烯及其衍生物,近年来非富勒烯受体材料也得到了迅速发展[10-12].电子给体材料则是由电子给体单元(donor)和电子受体单元(acceptor)共聚形成的D-A 共聚物.D-A共聚物具有很强的可设计性.由于D单元和A单元间的推拉电子作用,产生了分子内的电荷转移[13].D-A共聚物的HOMO能级主要取决于给体单元的HOMO能级,LUMO能级主要由受体单元的LUMO能级所决定[14].无论是富勒烯还是非富勒烯电池,其中的D-A共聚物给体材料均可通过对D/A单元的选取来优化电池的能级结构,提高材料的能级匹配度,使效率得到很大改善[12,14-17].
文献 [8,16]采用第一性原理密度泛函理论(density functional theory, DFT),通过对材料分子结构的优化设计,计算其电子结构和光学性质.其中文献[16]借助DFT方法,以二噻吩取代的苯并二噻吩(BDT)为给体单元, 具有双极性的吡咯并吡咯二酮(DPP)为受体单元,计算得到该共聚物的带隙值为3.89 eV.文献[8]利用 DFT方法,以苯并噻二唑(BT)为受体单元,苯并三噻吩(BTT)为给体单元,对给体单元进行优化设计后得出了5种同分异构体,并分别与同一个受体单元共聚.计算结果表明,5种聚合物的主链显示出不同的共面性,其2个相邻环间的二面角在12.3°~79.0°变化,聚合物主链的平面性越好,带隙值越小,其相应吸收谱的峰值也越强.可以看出,DFT在研究聚合物分子结构与电子结构时可以得到很有价值的结论[9-10].但是大量研究结果也表明,计算所得的聚合物带隙与实验值均有误差.研究聚合物材料时DFT所使用的理论模型和方法是否足够精确和可靠的问题研究目前还少有报道.本文基于DFT研究带隙的计算精度,针对不同类型密度泛函交换关联项的不同,采用3种泛函PBE、HSE06、B3LYP计算了大量的有机共轭聚合物,研究在DFT框架下聚合物的带隙和吸收光谱,考察了不同泛函对其精度的影响,所得结果具有一定的参考价值.
1 计算方法
采用基于第一性原理DFT的VASP软件包进行计算[18],交换关联泛函采用基于广义梯度近似的 PBE泛函、包含精确交换项的HSE06和B3LYP杂化泛函.PBE交换关联泛函考虑了电荷密度分布的不均匀性,包含一些空间变化的信息,能更加灵活地描述实际材料.B3LYP和HSE06属于杂化泛函,这2种泛函中都加入了精确交换项,其中B3LYP泛函精确交换项数值参数是由大量的计算数据拟合得到的,其泛函计算结果最接近实验值,但是采用B3LYP泛函计算很耗费机时.
采用投影缀加波 (PAW)方法来描述芯电子和价电子的相互作用.价电子波函数采用平面波展开,平面波截断动能为 400 eV,自洽场迭代能量精度为 1×10-4eV.结构优化过程中对所有原子进行完全弛豫,平均每个原子上力的收敛精度为0.2 eV/nm.布里渊区积分K点网格采用Monkhost-Pack方法产生,K点网格大小为9×1×1.
在计算过程中,用PBE、HSE06、B3LYP泛函分别计算了单噻吩(Th)及6种共轭聚合物的能级、带隙值和吸收光谱.这6种聚合物是以BDT为给体单元,与6种不同的受体单元形成的共聚物(PBDTTT、PBDTBT、PBDTNT、PBDTTTZ、PBDTPT、PBDTDPP).由于D-A共聚物侧链对共聚物的带隙和HOMO、LUMO能级影响较小,通常小于0.3 eV[19],同时为了结构优化和计算的方便,对所有结构单元的建模和计算都只保留主链结构,研究表明在计算聚合物的规律性问题时结果不受影响[13].
2 结果与讨论
2.1 分子结构模型中分子单元的个数对计算结果的影响
计算模型的建立取决于所用的软件.例如,基于DFT的 VASP软件包是以平面波为基组,以自由电子气为本征函数,计算模型是周期性结构,适合于计算长链结构聚合物的带隙.基于DFT的Gaussian软件是以原子轨道线性组合为基组,其解析基组为高斯函数,适合于计算低聚物小分子的带隙,也可以利用周期边界条件计算周期性体系.本文对VASP软件和Gaussian软件的计算结果进行了比较.
图1给出了VASP软件和Gaussian软件计算得到的能级图.如图1(a)所示,采用VASP软件计算了Th聚合物选取不同单元个数时的带隙值,其中计算模型是周期性结构.建模过程中在各原胞间加上合适的真空层(单个原胞间没有真空层时计算的是周期性结构),真空层的尺寸大于分子间相互作用的距离,各原胞中分子单元的个数分别为1~10.计算结果表明,随着原胞中分子单元个数的增加,带隙值逐渐减小,且趋近于周期性结构的值.此外,有限体系的带隙值收敛于无限周期的带隙值.

图1 VASP软件和Gaussian软件计算得到的能级图Fig.1 Energy levels obtained by using VASP sofeware and Gaussian software
根据文献[20],运用Gaussian软件得出4种聚合物(BDT-TP)n、(BDT-TD)n、(BDT-DTP)n、(BDT-TTP)n选取不同单元个数时的能级值,结果如图1(b)所示.可以看出,随着计算模型中单元个数的增加,带隙值也逐渐趋近于周期性结构的值.
从图1可以看出,VASP软件主要适用于周期性结构,建模过程中,如果在聚合物的各原胞间加上合适的真空层也可以计算小分子体系.Gaussian软件主要适用于低聚物小分子,加上周期性边界条件后,也可以用来计算周期性结构.同时,无论是VASP软件还是Gaussian软件,尽管计算过程中建模细节不同,计算函数基组不同,所计算的带隙值也有差别,但是对规律性问题的研究结果是一致的.
2.2 不同泛函对聚合物带隙值及吸收光谱的研究
DFT计算的基础就是对Khan-Sham方程进行求解,在得出的能量表达式中,动能、势能和库仑相互作用能都有准确的函数表达形式.而交换相关项包含许多非经典项,不能通过函数形式准确地表达.因此,产生了各种各样的密度泛函的近似表达.例如,局域密度近似泛函(LDA)、广义梯度近似(GGA)下的PBE泛函、杂化广义梯度近似(Hybrid-GGA)下的B3LYP泛函和以PBE为基础的HSE06泛函等.其中GGA下的PBE 泛函中,交换关联泛函是用局域电荷密度和电荷密度梯度[19]来表示的.B3LYP和HSE06属于杂化泛函,杂化是指在DFT中运用Hartree-Fock(HF)方法,并把HF中的准确交换作用通过杂化作用到精确交换项(EXC)中.杂化泛函的交换关联能包含了一定比例HF的交换关联项和一定比例DFT的交换关联项.B3LYP泛函的交换相关能为
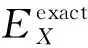
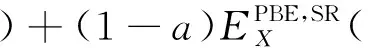
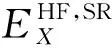
6种聚合物(PBDTTT、PBDTNT、PBDTBT、PBDTPT、PBDTTTZ、PBDTDPP)单体的分子结构如图2所示.分别使用PBE、HSE06、B3LYP泛函对Th以及6种聚合物进行了计算,不同泛函得出的理论带隙值如图3所示,并在图3中列出相应的实验值[21-26].PBDTNT、PBDTPT、PBDTTTZ和PBDTDPP的实验值是在聚合物中间加上π(噻吩)桥的值(文献[12]结果表明,在聚合物中间加上π桥其带隙值减小,与不加π桥的差别约为0.02 eV).
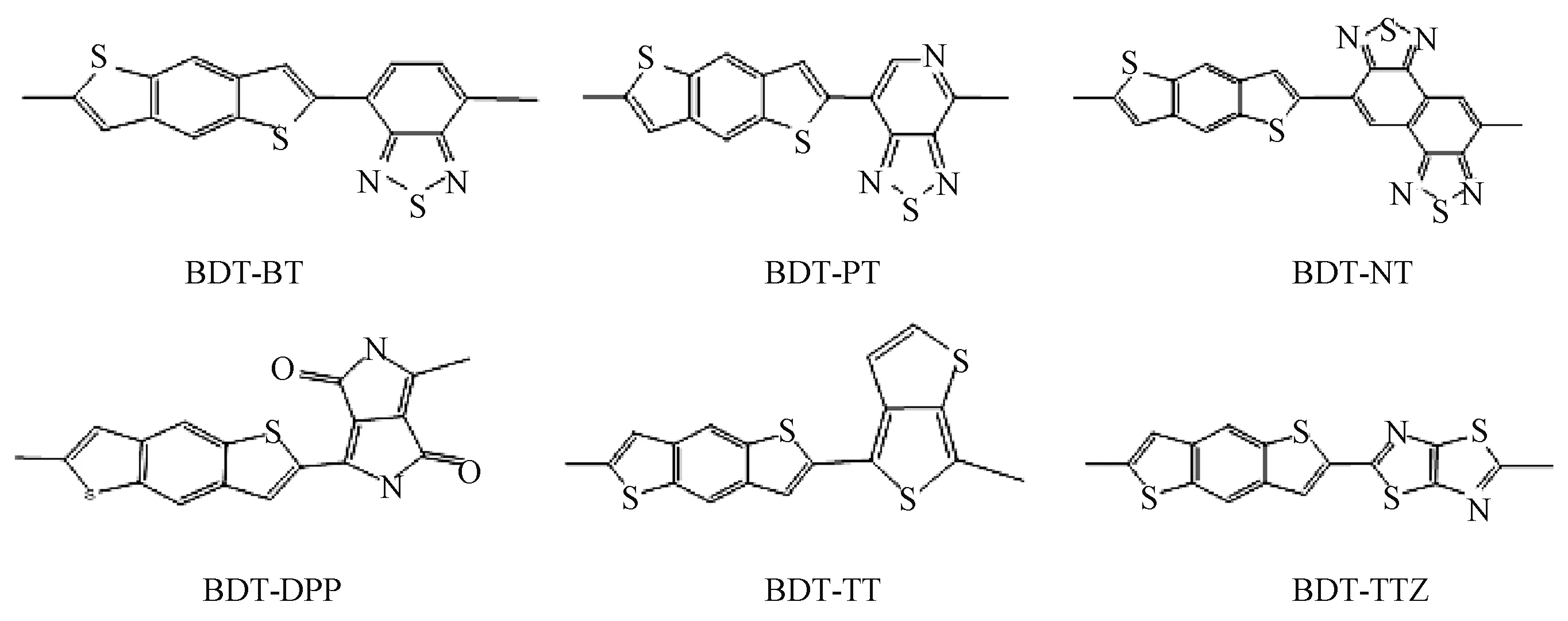
图2 6种聚合物单体的分子结构Fig.2 Monomer molecular structure of six polymers
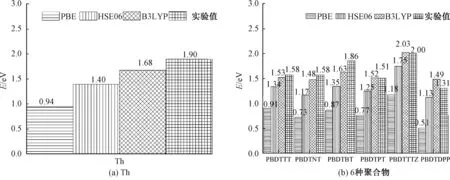
图3 PBE、HSE06、B3LYP泛函得出的Th和6种聚合物的理论带隙值Fig.3 Theoretical bandgap values of Th and six polymers obtained by PBE, HSE06, B3LYP functional
由图3可以看出,对于PBE泛函,其计算出的理论值远远小于其实验值,而B3LYP泛函是最接近实验值的,HSE06计算得出的带隙值介于PBE与B3LYP之间.PBE泛函是一个纯的GGA泛函,不包含eX项,B3LYP泛函含有20%的eX项,HSE06泛函在短程部分含有25%的eX项.计算结果的精确性与不同泛函中的精确交换项有关,这是因为共轭聚合物或者长链低聚物的许多重要性能都依赖于沿着共轭骨架的电子共轭度和轨道的物理离域程度.同时,不同泛函还具有不同的离域化误差特性.其中PBE具有明显的离域化误差特性,这使得其在计算中常常低估光学带隙值,B3LYP的离域化误差特性较PBE和HSE06要小.本文得出的带隙值结果与已有泛函的离域化误差特性的结论是一致的.
图4给出了给、受体单元和6种聚合物的能级图(按HOMO能级降低的顺序).如图4(a)所示,用PBE泛函计算了D单元和A单元的能级.D单元为BDT,A单元是TT、BT、NT、TTZ、PT、DPP.如图4(b)所示,用PBE、HSE06、B3LYP泛函分别计算了6种聚合物的能级.结果表明:① 聚合物的HOMO能级主要受D单元HOMO能级的影响, LUMO能级主要受A单元LUMO能级的影响;共聚物按HOMO能级降低的顺序排列的能级图也是一致的,不同泛函计算的结论一致;② 3种泛函计算的聚合物的带隙值均依次增加,B3LYP泛函计算的带隙值最大;③ B3LYP泛函计算的6种聚合物的带隙值和实验值一致.总之,不同泛函在研究规律性问题时结论是一致的.关于聚合物的带隙值,B3LYP泛函计算的结果和实验值最为接近.

图4 给、受体单元和6种聚合物的能级图Fig.4 Energy levels of the donor,acceptor and the six polymers
图5给出了PBDTPT不同泛函的吸收光谱和6种聚合物的B3LYP泛函吸收光谱.图5(a)为PBDTPT使用3种不同泛函得到的吸收谱,可以看出,当使用不同的泛函进行计算时,其在可见光区最高吸收峰的峰值会发生改变.对于PBE、HSE06、B3LYP泛函,其在可见光区的峰值分别为567.44、 432.55、 716.69 nm,图中标记的为B3LYP的实验值[24].可以看出,当使用B3LYP泛函对吸收谱进行研究时,峰值是最接近实验值的.
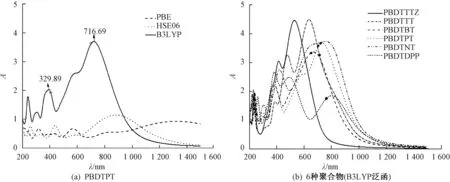
图5 PBDTPT不同泛函的吸收光谱和6种聚合物的B3LYP泛函吸收光谱Fig.5 Different functional absorption spectra of PBDTPT and B3LYP functional absorption spectra of six polymers
图5(b)为使用B3LYP泛函得出的6种聚合物的吸收光谱.图中标记处均为实验得出的最高吸收峰值[21-26].6种聚合物在可见近红外区的吸收波长范围分别为:PBDTTTZ(319.15~698.84 nm)、PBDTBT(346.72~874.88 nm)、PBDTTT(361.83~910.17 nm)、PBDTPT(341.68~945.44 nm)、PBDTDPP(314.11~942.77 nm)、PBDTNT(344.35~980.71 nm),对应的吸收波长宽度分别为370.69、528.16、548.34、603.76、628.66、636.36 nm,可以得出其在可见近红外区吸收波的吸收宽度由小到大的顺序为PBDTTTZ、PBDTBT、PBDTTT、PBDTPT、PBDTDPP、PBDTNT,并且其带隙值分别为2.03、1.63、1.53、1.52、1.49、1.48 eV,与带隙值逐渐减小的顺序一致.
3 结论
计算了Th共聚物分子结构模型中分子单元的个数对带隙值的影响,结果表明,无论是VASP软件还是Gaussian软件,尽管计算过程中建模细节不同,计算函数基组不同,所计算的带隙值也有差别,但是对规律性问题的研究结果是一致的.采用PBE、HSE06、B3LYP泛函计算6种聚合物(PBDTTT、PBDTBT、PBDTNT、PBDTTTZ、PBDTPT、PBDTDPP)的带隙值及吸收光谱,结果表明,PBE、HSE06、B3LYP泛函计算的聚合物的带隙值均依次增加;B3LYP泛函计算的带隙值最接近实验值;对于共轭聚合物性能的研究,不管使用何种泛函,在研究规律性问题时结果是一致的.
猜你喜欢
广西大学学报(自然科学版)(2022年4期)2022-09-19
无机化学学报(2022年9期)2022-09-16
东华大学学报(自然科学版)(2022年3期)2022-06-25
井冈山大学学报(自然科学版)(2022年2期)2022-03-31
商品与质量(2020年18期)2020-07-27
科技创新与应用(2018年21期)2018-09-14
振动工程学报(2017年4期)2018-05-31
电子制作(2018年1期)2018-04-04
振动工程学报(2017年1期)2017-04-21
读写算·教研版(2016年8期)2016-05-07
