
儿童新诊断与慢性免疫性血小板减少症差异基因分析
2019-02-18 07:37马利敏杨学文杨海平阮林海
中国医药生物技术 2019年1期
马利敏,杨学文,杨海平,阮林海
儿童新诊断与慢性免疫性血小板减少症差异基因分析
马利敏,杨学文,杨海平,阮林海
471023 洛阳,河南科技大学第一附属医院血液科
筛选儿童新诊断与慢性免疫性血小板减少症(ITP)之间的差异表达基因(DEGs)并进行生物信息学分析。
从基因表达数据库中下载芯片表达谱 GSE46922 数据集,利用 BRB-ArrayTools 软件鉴定 DEGs,然后分别对差异基因进行基因本体(GO)功能富集分析、Pathway 富集分析和互作网络分析。
共筛选出 1225 个 DEGs,其中上调基因 665 个,下调基因 560 个。GO 富集分析发现 DEGs 主要参与转录调控、小分子代谢、蛋白泛素化、凋亡调控、固有免疫反应、病毒复制等生物学过程。Pathway 富集分析发现 DEGs 显著富集于代谢通路、内质网蛋白加工、破骨细胞分化、MAPK 信号通路、病毒感染、凋亡等。网络分析鉴定出的核心基因有 CHD4、UQCR10、AP2M1、SIRPγ 和 GPR180,核心Pathways 包括 MAPK 信号通路、细胞周期和细胞凋亡。
明确了儿童新诊断与慢性 ITP 的基因表达谱不同,为进一步阐明儿童 ITP 发生发展的分子机制和指导早期治疗干预提供了基础。
血小板减少症; 基因表达谱; 儿童; 计算生物学; 差异表达基因; 免疫应答
原发性免疫性血小板减少症(immune thrombocytopenia,ITP)是儿童最常见的出血性疾病,主要表现为急性起病,皮肤黏膜出现出血点和瘀斑,外周血循环中血小板计数低于正常伴骨髓巨核细胞发育成熟障碍。绝大多数患儿为急性发病,呈自限性疾病,病程小于 3 个月,定义为新诊断 ITP,预后良好。但有 20% ~ 30% 的患儿可发展为慢性 ITP,病程迁延反复,持续 12 个月以上,严重影响患儿的生活质量[1]。研究表明,免疫调节功能紊乱、遗传背景或继发性因素导致血小板破坏增加和生成减少在 ITP 的发病中起着重要作用,但具体机制尚不完全清楚[2-3]。目前尚未发现有效的分子标志物在临床上区分新诊断 ITP 和慢性 ITP,也无法准确预测新诊断 ITP 患儿的长期结局。本研究利用基因表达公共数据库(gene expression omnibus,GEO)中儿童 ITP 相关芯片数据,对儿童新诊断和慢性 ITP 的基因表达谱进行比较和分析,探索新诊断 ITP 与慢性 ITP 分子特征的异同,为进一步阐明儿童 ITP 发生发展的分子机制和指导早期干预提供基础。
1 资料与方法
1.1 资料来源
从 GEO 数据库中下载芯片表达谱 GSE46922数据集,该数据集由 Jernås 等[4]提交,包含了 7 个新诊断 ITP(GSM1141222-GSM1141228)和 6 个慢性 ITP(GSM1141229-GSM1141234)患儿外周血 T 细胞的表达谱数据。实验设计为收集新诊断 ITP 和慢性 ITP 患儿的肝素抗凝外周血,立即采用密度梯度离心法分离外周血单个核细胞,去除CD14+细胞后,利用 CD3+磁珠收集 T 细胞,–80 ℃保存,然后提取总 RNA 并检测其浓度和纯度。采用 GPL570 平台,即 Affymetrix Human Genome U133 Plus 2.0 Array 基因芯片进行检测。
1.2 方法
1.2.1 差异表达基因筛选 将 GSE46922 原始芯片数据导入 BRB-ArrayTools 软件包,先进行芯片信号值的预处理和标准化,然后以慢性 ITP芯片表达谱为对照组,新诊断 ITP 芯片表达谱为实验组,通过基因芯片显著性分析(significant analysis of microarray,SAM)方法计算每个基因的统计学显著水平值与误判率值,差异表达基因(differentially expressed genes,DEGs)的筛选标准为:< 0.05,< 0.05,差异倍数≥ 1.5 倍。同时计算基因与样本间的相关性,按照先基因、后样本的顺序将差异基因的表达数据进行层次聚类分析[5]。
1.2.2 GO 功能富集分析 基因本体(gene ontology,GO)是基因功能的标准化分类系统,用于限定和描述基因及其蛋白功能。本研究基于 GO 数据库,利用 Fisher 检验统计 GO 富集各条目的概率值,同时用 Benjamini-Hochberg 法进行校正以控制错误发现率(false discovery rate,FDR),显著富集 GO 条目的标准为< 0.05,FDR < 0.05。
1.2.3 Pathway 富集分析 KEGG 数据库有助于把基因及其表达情况以网络的形式进行研究,系统分析基因的功能以及基因组之间的信息,注解生物系统的高级功能[6]。本研究基于 KEGG 数据库,采用 Fisher 检验统计DGEs 在 Pathway 中的富集程度,< 0.05 和 FDR < 0.05 的 Pathway 定义为显著富集的 Pathway。
1.2.4 互作网络分析 根据图论的原理,以 Pathway 为研究对象,将显著性富集的 Pathways 基于 KEGG 数据库中的相互作用关系构建 Pathway 相互作用网络[7]。对输入的差异基因表达谱数据,首先计算出各基因间的相关系数矩阵、各节点间的连接度和不相似度,再通过基因间的相互关系拟合基因的无标度网络关系,从而得到加权基因共表达网络[8]。
2 结果
2.1 差异基因筛选结果
以慢性 ITP 为对照组,新诊断 ITP 为实验组,根据筛选标准从 GSE46922 数据集中共筛选出 1225 个 DGEs,其中包含 665 个上调表达基因,560 个下调表达基因,部分差异基因列于表 1。
2.2 GO 功能富集分析
通过 GO 分类系统对 DGEs 进行注释后发现,显著富集的分子功能类别共有 82 个 GO 条目,主要包括蛋白结合、ATP 结合、离子结合、蛋白激酶活性和核酸结合等;显著富集的生物过程类别共涉及 192 个 GO 条目,主要包括转录调控、小分子代谢、蛋白泛素化、凋亡调控、固有免疫反应、病毒复制、血液凝固、Toll 样受体(toll-like receptor,TLR)通路、细胞自噬等,生物过程类别富集水平最显著的前 20 个 GO 条目列于表 2。
2.3 Pathway 富集分析
Pathway 富集分析有助于明确 DGEs 所参与的主要代谢途径和细胞转导通路,结果发现这些差异基因显著富集于代谢通路、内质网蛋白加工、破骨细胞分化、丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)信号通路、病毒感染、凋亡、T 细胞受体通路等 88 个相关通路,其中富集水平最显著的前20 条通路列于表 3。
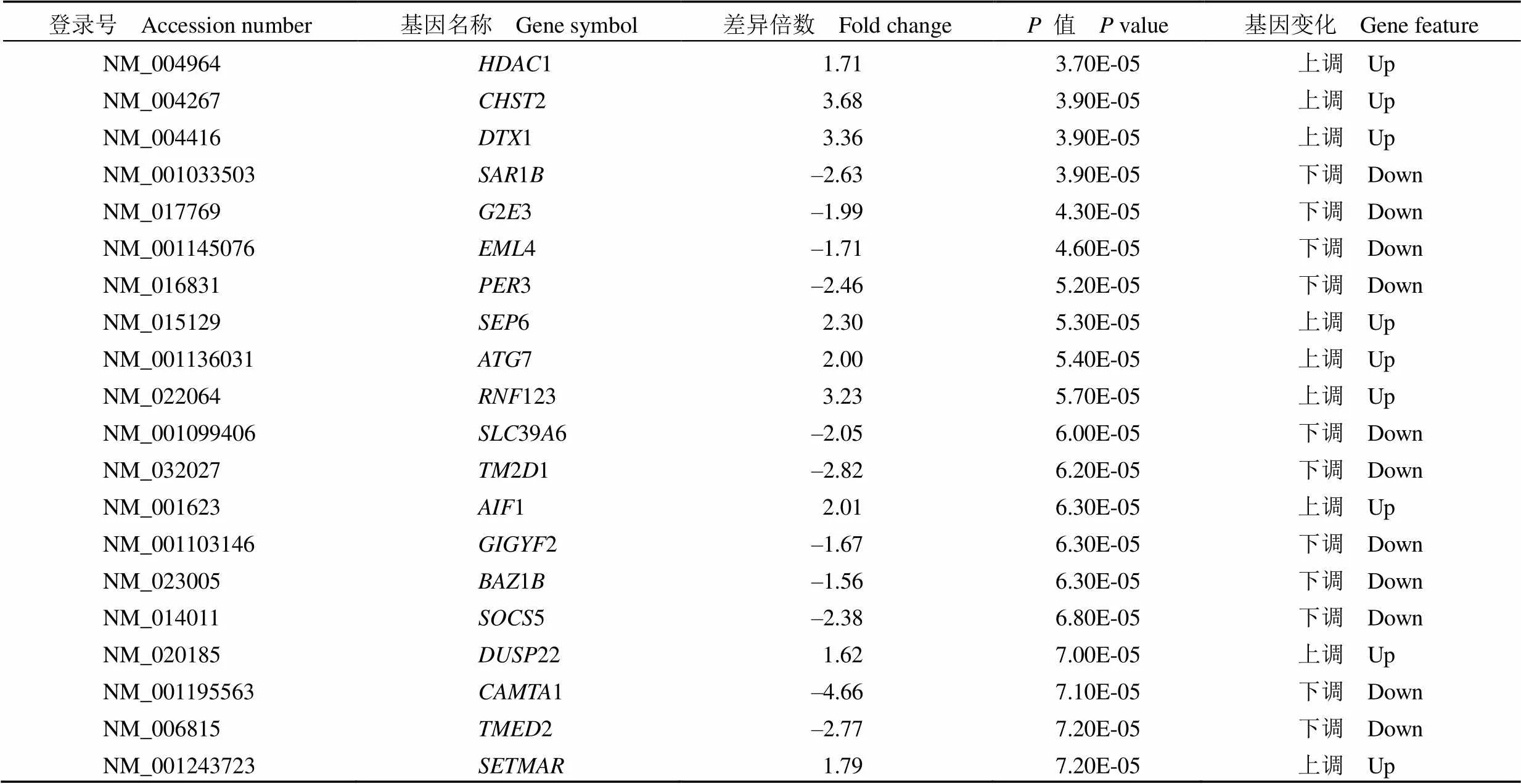
表 1 部分差异表达基因列表

表 2 差异表达基因 GO 富集分析
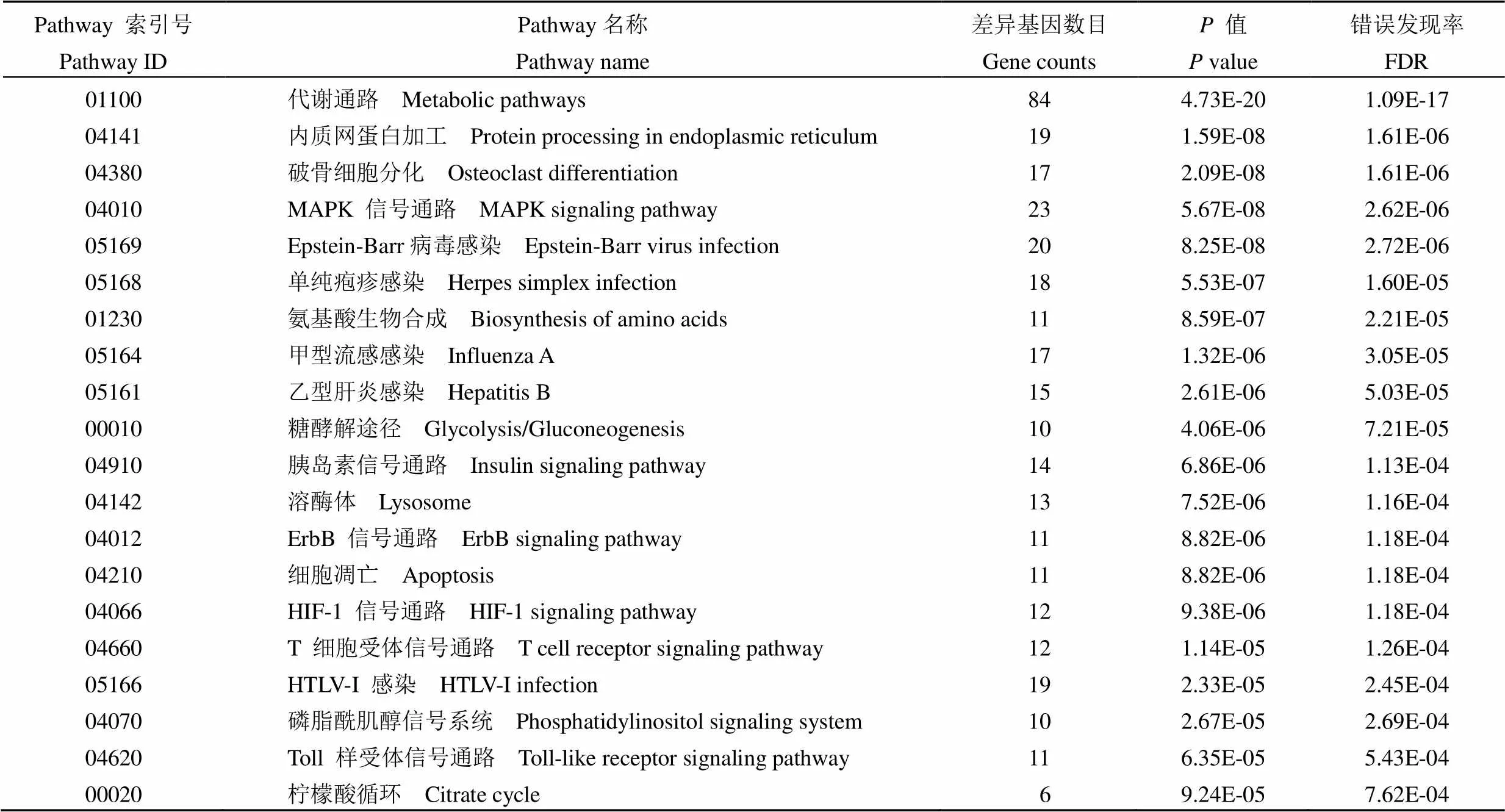
表 3 差异表达基因 Pathway 富集分析
2.4 互作网络分析
基于 KEGG 数据库中信号通路的上下游关系,构建显著性 Pathways 之间的相互作用网络如图 1 所示,圆点表示 Pathway,带箭头的实线表示 Pathway 的上下游关系。圆点越大,表明与之有上下游关系的 Pathway 数目越多,在网络中的作用也越重要,核心 Pathways 包括 MAPK 信号通路、细胞凋亡和细胞周期。基于差异基因表达谱数据,构建的基因共表达网络发现核心基因包括染色质域解旋酶 DNA 结合蛋白 4(chromodomain helicase DNA binding protein 4,CHD4)、泛醇细胞色素 C 还原酶复合物 III 亚基 10(ubiquinol-cytochrome C reductase, complex III subunit X,UQCR10)、衔接因子相关蛋白复合体2 μl(adaptor-related protein complex 2 Mu 1,AP2M1)、信号调节蛋白 γ(signal-regulatory protein γ,SIRPγ)、G 蛋白偶联受体 180(G protein-coupled receptor 180,GPR180)。
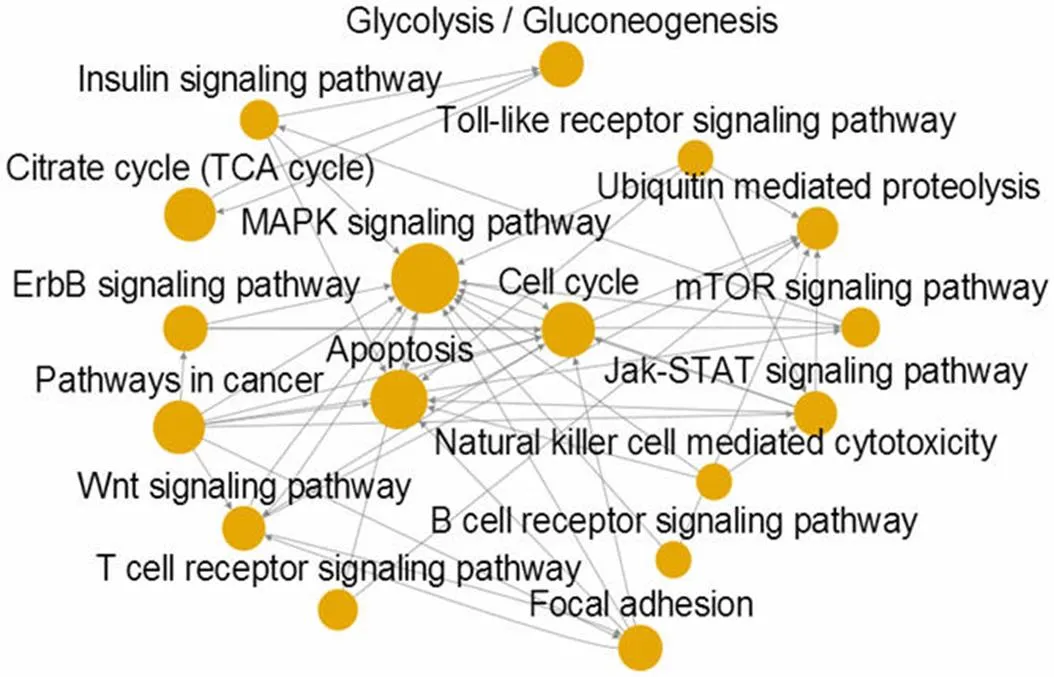
图 1 Pathway 相互作用网络分析
Figure 1 The network analysis of Pathway interactions
3 讨论
通常认为 ITP 的发病基于一定的遗传物质基础,在某些诱因下的机体免疫功能紊乱,特别是 B 细胞和 T 细胞调节功能的失调。免疫调节功能异常,B 细胞过度激活,产生抗血小板自身抗体,使血小板在网状内皮系统破坏。ITP 患儿的血小板被自身抗体致敏后,单核巨噬细胞系统通过结合暴露的抗体 Fc 段,吞噬致敏血小板,而 T 细胞功能异常使 B 细胞异常活化产生血小板表面相关抗原抗体,加速血小板的破坏。近年来研究发现 T 细胞及其亚群的异常可能是 ITP 免疫发病机制的关键[9-10]。
本研究利用 GEO 数据库中儿童 ITP 相关芯片数据,对儿童新诊断和慢性 ITP 的基因表达 谱进行比较,共筛选出 1225 个差异基因,包括 665 个上调基因和 560 个下调基因,其中1 和1分别在上调和下调基因中差异最为显著。1 参与基因表达的表观遗传学调控、机体内病毒转录抑制、细胞周期、血液凝固和 EB 病毒感染等过程,在多种肿瘤和免疫相关性疾病中发挥重要作用;1具有 GTP 酶活性,主要参与细胞内蛋白转运、囊泡介导的胞内运输和抗原处理与提呈过程,这两个基因在 ITP 发病及转归中的功能尚无报道。层次聚类分析发现新诊断 ITP 患儿与慢性 ITP 患儿的表达谱在聚类树形图中没有出现重叠,表明新诊断 ITP 与慢性 ITP 的分子特征不同。GO 功能富集和通路富集分析结果发现这些差异基因主要参与转录调控、小分子代谢、蛋白泛素化、凋亡调控、固有免疫反应、病毒复制、血液凝固、TLR 通路、细胞自噬等生物学过程,主要富集于代谢通路、破骨细胞分化、MAPK 信号通路、病毒感染、凋亡、T 细胞受体通路等信号通路。参与 T 细胞活化、淋巴细胞增殖、TLR 信号、趋化等过程的基因在新诊断 ITP 中高表达,而涉及 B 细胞分化、CD8+α:β T 细胞谱系定型等过程的基因在慢性 ITP 中高表达,这表明新诊断 ITP 涉及更加广泛、非特异的炎症基因活化,而慢性 ITP 主要是 Th1 免疫应答,自身抗体产生和 CD8+细胞毒性 T 细胞。
TLRs 通过识别并结合病原相关分子模式,激活信号传导通路,调节免疫应答,增强吞噬细胞吞噬能力和免疫细胞杀伤能力,TLR2 和 TLR4 在慢性 ITP 患儿中表达下降,使其病毒清除能力和免疫应答能力下降,而白细胞介素 2(interleukin 2,IL-2)、IL-10、γ 干扰素在慢性 ITP 患者血清中增高,进而激活巨噬细胞、细胞毒性 T 细胞、补体形成,吞噬、破坏血小板[11-12]。IL-4 可促进 B 细胞活化、增殖分化和产生抗体,本研究分析发现 IL-4 在慢性 ITP 患儿 T 细胞中高表达。马静瑶 等[13]对初诊 ITP 患儿的 T 辅助细胞因子进行检测,发现治疗前 IL-4 和干扰素水平升高可能是导致 ITP 病程迁延的因素之一,尤其 IL-4 增高更明显,提示 IL-4 可作为 ITP 患儿在初次发病时判断预后的指标之一。另外本研究发现 IL-16 在新诊断 ITP 患儿外周血 T 细胞中的表达水平高于慢性 ITP 患儿,而且网络分析发现 IL-16 在基因共表达网络中发挥关键作用。Abd El-Glil 和 Assar[14]发现新诊断 ITP 患儿血清中 IL-16 水平比持续性和慢性 ITP 患儿高,持续性 ITP 患儿 IL-16 水平又比慢性 ITP 患儿高,而且 IL-16 水平与网织血小板和血小板计数呈负相关关系,提示 IL-16 可用于预测儿童 ITP 的临床病程。本研究发现 IL-16 在新诊断 ITP 患儿外周血 T 细胞中的表达水平高于慢性 ITP 患儿,而且 IL-16 在基因共表达网络中发挥关键作用。Yao 等[15]发现新诊断和慢性 ITP 患儿 CD4+T 细胞及 CD19+B 细胞中 Bcl-6、c-Maf、Blimp-1、ICOSL、TACI、BAFFR 的 mRNA 表达水平差异显著,这些因子的表达失衡引起滤泡性辅助性 T 细胞过度活化,进而可能导致儿童 ITP 的免疫发生和病程迁延。李珊珊等[16]研究发现,慢性 ITP 组膜糖蛋白 GP IX、GP III a、GMP140 特异性自身抗体明显高于健康对照组,但低于新诊断 ITP 组,表明血小板自身抗体在儿童慢性 ITP 病情迁延中可能发挥重要作用。本研究互作网络分析发现的核心基因包括 CHD4、UQCR10、AP2M1、SIRPγ 和 GPR180,如 SIRPγ 参与细胞黏附、血液凝固、T 细胞活化、白细胞迁移等过程,AP2M1 涉及防御反应、病毒感染、抗原提呈、表皮生长因子受体通路抑制等,这些核心基因及核心通路在新诊断 ITP 与慢性 ITP 差异表达的意义有待深入研究。
总之,通过对儿童新诊断和慢性 ITP 的基因表达谱进行比较和生物信息学分析,明确了新诊断 ITP 与慢性 ITP 分子特征不同,为进一步阐明儿童 ITP 发生发展的分子机制、筛选有效分子标志物和指导早期治疗干预提供基础。
[1] Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood, 2009, 113(11):2386-2393.
[2] Cines DB, Cuker A, Semple JW. Pathogenesis of immune thrombocytopenia. Presse Med, 2014, 43(4 Pt 2):e49-e59.
[3] Wang TY, Wu RH. New progresses in diagnosis and treatment of childhood immune thrombocytopenia--open the new era based on immunologic pathogenesis. J Appl Clin Pediatr, 2016, 31(15):1121- 1125. (in Chinese)
王天有, 吴润晖. 开启建立在免疫发病机制上的儿童免疫性血小板减少症诊疗新时代. 中华实用儿科临床杂志, 2016, 31(15):1121- 1125.
[4] Jernås M, Hou Y, Strömberg Célind F, et al. Differences in gene expression and cytokine levels between newly diagnosed and chronic pediatric ITP. Blood, 2013, 122(10):1789-1792.
[5] Li J, Fan S, Han D, et al. Microarray gene expression profiling and bioinformatics analysis of premature ovarian failure in a rat model. Exp Mol Pathol, 2014, 97(3):535-541.
[6] Kanehisa M, Goto S, Kawashima S, et al. The KEGG resource for deciphering the genome. Nucleic Acids Res, 2004, 32(Database issue): D277-D280.
[7] Zhuang Q, Mao W, Xu P, et al. Identification of differential genes expression profiles and pathways of bone marrow mesenchymal stem cells of adolescent idiopathic scoliosis patients by microarray and integrated gene network analysis. Spine (Phila Pa 1976), 2016, 41(10): 840-855.
[8] Li Y, Li Y, Bai Z, et al. Identification of potential transcriptomic markers in developing pediatric sepsis: a weighted gene co-expression network analysis and a case-control validation study. J Transl Med, 2017, 15(1):254.
[9] Wang YC, Liu MJ, Zhu GY, et al. Significance of Th17/Treg imbalance in children with primary immune thrombocytopenia. Chin J Contemp Pediatr, 2016, 18(3):238-242. (in Chinese)
王颖超, 刘满菊, 朱桂英, 等. Th17/Treg细胞比例失衡在儿童原发性免疫性血小板减少症中的意义. 中国当代儿科杂志, 2016, 18(3): 238-242.
[10] El-Rashedi FH, El-Hawy MA, Helwa MA, et al. Study of CD4+, CD8+, and natural killer cells (CD16+, CD56+) in children with immune thrombocytopenic purpura. Hematol Oncol Stem Cell Ther, 2017, 10(1):8-14.
[11] Wang CM, Sheng GY, Zou X, et al. Levels of toll-like receptors-2, -4 on platelets in children with idiopathic thrombocytopenic purpura. Chin J Contemp Pediatr, 2009, 11(10):797-801. (in Chinese)
王春美, 盛光耀, 邹湘, 等. 血小板表面TLR2和TLR4在儿童特发性血小板减少性紫癜中的变化及意义. 中国当代儿科杂志, 2009, 11(10):797-801.
[12] Bennett CM, Tarantino M. Chronic immune thrombocytopenia in children: epidemiology and clinical presentation. Hematol Oncol Clin North Am, 2009, 23(6):1223-1238.
[13] Ma JY, Wu RH, Chen ZP, et al. Initial study of T-helper cell cytokines level in children patient with primary immunne thrombocytopenia during the initial episode. Chin J Thromb Hemost, 2013, 19(3):110- 114, 117. (in Chinese)
马静瑶, 吴润晖, 陈振萍, 等. 儿童原发性免疫性血小板减少症初发病例T辅助细胞因子表达水平意义初探. 血栓与止血学, 2013, 19(3):110-114, 117.
[14] Abd El-Glil RR, Assar EH. Level of IL-16 and reticulated platelets percentage during the clinical course of immune thrombocytopenic purpura in children. Egypt J Immunol, 2015, 22(1):29-40.
[15] Yao X, Li C, Yang J, et al. Differences in frequency and regulation of T follicular helper cells between newly diagnosed and chronic pediatric immune thrombocytopenia. Blood Cells Mol Dis, 2016, 61: 26-36.
[16] Li SS, Jiang H, Xia M. Expression of immune autoantibodies in children with persistent/chronic immune thrombocytopenia. J Appl Clin Pediatr, 2015, 30(7):517-520. (in Chinese)
李珊珊, 蒋慧, 夏敏. 持续性和慢性免疫性血小板减少症患儿免疫抗体表达的意义. 中华实用儿科临床杂志, 2015, 30(7):517-520.
Analysis of differential genes between newly diagnosed and chronic pediatric immune thrombocytopenia
MA Li-min, YANG Xue-wen, YANG Hai-ping, RUAN Lin-hai
Department of Hematology, The First Affiliated Hospital, College of Clinical Medicine of Henan University of Science and Technology, Luoyang 471023, China
To screen and analyze the differentially expressed genes (DEGs) between newly diagnosed and chronic pediatric immune thrombocytopenia (ITP) in bioinformatic database.
The microarray expression profiles dataset GSE46922 was downloaded from the Gene Expression Omnibus database. The BRB-ArrayTools software was applied to control quality and identify the DEGs. Then the Gene Ontology (GO) function enrichment, Pathway enrichment, gene co-expression network, and pathway interaction network analysis were performed based on these differential genes.
Total 1225 DEGs were screened out, of which 665 genes were up-regulated and 560 genes were down-regulated. GO enrichment analysis showed that DEGs mainly participated in the biologic processes of transcription regulation, small molecule metabolic process, protein ubiquitination, apoptotic process, innate immune response and viral reproduction. Pathway enrichment analysis suggested that DEGs were enriched in metabolic pathways, protein processing in endoplasmic reticulum, osteoclast differentiation, MAPK signaling pathway, virus infection, apoptosis, etc. The network analyses identified the hub genes such as CHD4, UQCR10, AP2M1, SIRPγ, GPR180, and core pathways including MAPK signaling pathway, cell cycle and apoptosis.
The gene expression profiles differ between newly diagnosed and chronic pediatric ITP. The analysis provides the foundation for clarifying the molecular mechanism underlying the pathogenesis and development of ITP and for guiding early therapeutic intervention for pediatric ITP.
Thrombocytopenia; Gene expression profiling; Child; Computational biology; Differentially expressed genes; Immune response
YANG Hai-ping, Email: yfyyanghaiping@163.com
洛阳市科技计划项目(1503007A-4)
杨海平,Email:yfyyanghaiping@163.com
2018-10-19
10.3969/j.issn.1673-713X.2019.01.007
猜你喜欢
人人健康(2022年13期)2022-07-25
中国水运(2022年4期)2022-04-27
建材发展导向(2022年4期)2022-03-16
卫星应用(2022年1期)2022-03-09
安徽医专学报(2020年6期)2021-01-15
世界最新医学信息文摘(2020年68期)2020-08-28
临床医药文献杂志(电子版)(2020年89期)2020-04-27
心理学报(2020年1期)2020-02-27
心电与循环(2020年1期)2020-02-27
江苏农业科学(2017年5期)2017-04-15
