
从雄烯二酮发酵废液中分离甾醇和油脂的工艺研究
2019-02-15 09:23刘凤霞娄源民何际芳
中国粮油学报 2019年1期
王 莹 刘凤霞 薛 刚 娄源民 何际芳
(南阳理工学院生物与化学工程学院1,南阳 473004)(河南省工业微生物资源与发酵技术重点实验室2,南阳 473004)(河南恒天久大实业有限公司3,郑州 450001)
雄烯二酮在甾体激素类药物中是不可或缺的重要中间体,现代工业主要采用双液相体系甾体微生物转换法生产雄烯二酮[1-2]。微生物转化甾醇的培养系统需要添加20%的植物油和0.4%的甾醇,经微生物转化、甲醇提取、脱色、浓缩、结晶、重结晶得合格的雄烯二酮[3-6]。回收甲醇后的乳化液中含有70%~80%油脂和没被转化的甾醇,且含有较高浓度油脂和微生物菌体、蛋白质、多糖等物质,最终结合产生高度稳定的乳化液废弃物,其处理问题成为目前制约企业发展的一个难点[7-11]。
本研究探索一套合理的回收油脂和甾醇工艺,使回收的油脂经过初步精炼后能够循环使用,或者将回收油脂能够作为生物柴油的原料,并将回收的甾醇作为雄烯二酮生产的原料,研究雄烯二酮发酵废液的变废为宝及资源化综合利用,以期为其进一步开发利用提供参考。
1 材料与方法
1.1 材料与试剂
实验所用乙酸乙酯、石油醚、甲醇、三氯甲烷、正丁醇等试剂均为分析纯;雄烯二酮发酵废液由河南恒天久大实业有限公司提供。
1.2 仪器与设备
7890B气相色谱仪;GL-20G-C高速冷冻离心机;RE-52A旋转蒸发器。
1.3 甾醇与雄烯二酮的检测
样品处理:取发酵液中上层样品1~2 mL,用同样体积的乙酸乙酯充分溶解,静置分层,取上层清液稀释100倍后进样检测。
气相色谱检测条件:色谱柱mxt 5,柱温210 ℃,保持3 min,以30 ℃/min的速度升温到290 ℃保持10 min,进样口温度300 ℃,FID 300 ℃,载气分流比是20∶1,氮气流速1 mL/min,空气流速500 mL/min,进样量5 μL。
1.4 分离甾醇和油脂的工艺步骤
将雄烯二酮发酵废液进行预热处理,按一定比例加入萃取溶剂,经过在一定的温度条件下,进行萃取,经离心后,去沉淀,上清液放入一定温度条件的冰箱里,冷却结晶后,离心,沉淀物即为甾醇粗品,上清液进行减压蒸馏浓缩,回收溶剂,并得到副产品大豆油,在大豆油中加入溶解雄烯二酮效果较好的有机溶剂,同样进行萃取,离心后冷冻结晶,待结晶养成后,经离心得到雄烯二酮粗品。上清液再次浓缩,回收溶剂和副产品大豆油。分离甾醇和雄烯二酮工艺示意图如图1所示。

图1 分离甾醇和油脂工艺示意图
1.5 有机溶剂萃取法提取甾醇的工艺参数优化
1.5.1 有机溶剂的选择
称取雄烯二酮发酵废液5 g,分别按体积比为1∶3在试管加入有机溶剂甲醇、乙酸乙酯、石油醚、三氯甲烷、乙醇、丙酮、正丁醇各15 mL,并标记名称,置于50 ℃恒温水浴锅中2 h后,观察其分相情况以及现象,以萃取液上相高度为基准,确定最佳的有机溶剂。
1.5.2 料液比的选择
称取五份雄烯二酮发酵废液5 g,按1∶1、1 ∶1.5、1∶2、1∶2.5、1∶3的不同料液比加入最佳有机溶剂,置于恒温水浴50 ℃条件下2 h后,观察其分相情况,以萃取液上相溶液的高度为基准,确定最佳的料液比。平行实验,重复3次。
1.5.3 萃取温度的选择
称取四份雄烯二酮发酵废液5 g,选择确定的最佳料液比加入有机溶剂,分别在室温(19 ℃)、30、40、50 ℃条件下2 h后,观察其分相情况与现象,以萃取液上相溶液的高度为基准,确定最佳的萃取温度。平行实验,重复3次。
1.5.4 萃取时间的选择
称取3份雄烯二酮发酵废液5 g,选择确定的最佳萃取温度加入有机溶剂,分别在30、60、90、120 min的不同时间条件下,观察其分相情况与溶液的透明度,以萃取液上相溶液的高度为基准,确定最佳的萃取时间。平行实验,重复3次。
1.5.5 萃取次数的选择
分别称3份取雄烯二酮发酵废液5 g,分别萃取1、2、3次,在恒温水浴50 ℃下2 h,吸取上相溶液,在-10 ℃条件下,离心10 min,转速4 000 r/min,取上清液到试管中,放入0 ℃的冰箱中结晶6 h,然后过滤,称量沉淀物的湿重。平行实验,重复3次。
1.5.6 不同浓度有机溶剂对分离甾醇工艺的影响
分别称取4份雄烯二酮发酵废液5 g,按优选的最佳条件,分别加浓度为100%、96%、93%、90%的有机溶剂进行萃取,在恒温水浴50 ℃下萃取2 h,吸取上相溶液在-10 ℃离心10 min,转速4 000 r/min,并记录上相溶液体积。经离心后,取上清液倒入试管中,在0 ℃下结晶6 h,再次离心,称量沉淀物湿重。平行实验,重复3次。
1.6 冷却结晶法分离甾醇的工艺参数优化
1.6.1 结晶温度的选择
分别称取3份雄烯二酮发酵废液5 g,按实验确定的最佳萃取次数,在恒温水浴50 ℃下2 h后,经萃取后,取上相于-10 ℃离心10 min,转速4 000 r/min,取上清液倒入小试管中,并编号,在10、0、-10 ℃条件下冷却结晶6 h,观察其结晶情况与现象。然后过滤,称量沉淀物的湿重。平行实验,重复3次。
1.6.2 结晶次数的选择
分别称取3份雄烯二酮发酵废液5 g,在实验确定的最佳条件下,进行实验后,取萃取上相溶液于-10 ℃离心10 min,转速4 000 r/min,取上清液倒入小试管中,并编号,放在确定的最佳条件下,结晶6 h后,经过滤,沉淀物称量其湿重,而滤液再次倒入试管,放入冰箱继续结晶6 h,然后过滤,并再次称量其湿重,取滤液再次结晶6 h。再次过滤,称量沉淀物湿重。观察3次结晶的情况与现象。平行实验,重复3次。
1.7 溶剂回收及雄烯二酮的分离
将分离甾醇后的上清液进行减压蒸馏回收溶剂。并得到一定浓度的浓缩物,然后向浓缩物中添加溶剂甲醇进行萃取,浓缩甲醇相到一半,冷冻结晶,冷冻离心沉淀物即雄烯二酮及其他副产品。再将上清液进行浓缩回收有机溶剂以及其他副产品。
2 结果和分析
2.1 有机溶剂萃取法提取甾醇的工艺参数优化
2.1.1 有机溶剂的选择比较
称取质量5 g的雄烯二酮发酵废液,按料液比1∶3分别加入丙酮、甲醇、石油醚、三氯甲烷、乙酸乙酯、乙醇等有机溶剂15 mL,充分摇匀,在恒温水浴50 ℃下萃取2 h后,观察溶剂的萃取情况。结果显示:甲醇、乙醇、丙酮虽然能够快速分出沉淀,但得到的甾醇和油较少。石油醚和三氯甲烷则无法高效地实现破乳效果,影响后续工艺操作;只有乙酸乙酯作为萃取剂时,不会形成乳化液,因此破乳剂优选为乙酸乙酯。乙酸乙酯与乙醇最好,其余溶剂的效果不佳。乙酸乙酯作为中等极性溶剂,在萃取油水两相中不会形成乳化液,且较容易过滤。而其他非极性溶剂在分离时会形成乳化液,对甾醇的分离造成很大的影响,使得分离甾醇较难。因此,本实验初步选择的最佳溶剂为乙酸乙酯。
2.1.2 料液比对分离工艺的影响
称取5份雄烯二酮发酵废液5 g,按1∶1、1 ∶1.5、1∶2、1∶2.5、1∶3的不同料液比加入溶剂乙酸乙酯,置于恒温水浴50 ℃条件下2 h后,观察其分相情况以及萃取液的颜色透明度变化,以萃取液上相溶液的高度为基准,有机溶剂不同料液比的沉降高度比较见图2。
为判断不同料液比对分离甾醇工艺的影响是否显著,以确定最佳的料液比,对上述单因素进行方差分析,不同料液比对萃取量的影响方差分析见表1。
结果显示:料液比对萃取分离甾醇的结果达到极显著水平,为了确定不同料液比与分离效果的影响差异,采用LSD(最少显著数法)进行显著性比较。不同料液比对乙酸乙酯高度的显著性比较见表2。

图2 有机溶剂不同料液比、不同萃取时间及温度的乙酸乙酯相沉降高度比较

料液比平均数差异显著性α=0.05α=0.01萃取次数平均数差异显著性α=0.05α=0.011∶39.167aA2次1.267aA1∶2.57.567bB3次0.778bB1∶26.733cC1次0.633cC1∶1.54.033dD1∶13.433eE
由结果分析:在料液比在1∶1到1∶3之间,乙酸乙酯的萃取率在随料液比的增加不断增大,五组不同料液比均达到极显著水平。因此选取料液比为1∶3最佳。
2.1.3 萃取温度对分离工艺的影响
称取4份雄烯二酮发酵废液5 g,按料液比为1 ∶3加入15 mL的乙酸乙酯溶剂,分别在室温(19 ℃)、30、40、50 ℃条件下2 h后,观察其分相情况与现象,以萃取液上相溶液的高度为基准,不同萃取温度的分析比较见图2。为判断不同萃取温度对分离甾醇工艺的影响是否显著,以确定最佳的温度,对上述单因素进行方差分析,不同萃取温度对分离工艺的影响方差分析见表3。
结果显示,萃取温度对萃取分离甾醇的结果达到极显著水平,为了确定不同温度与分离效果的影响差异,采用LSD(最少显著数法)进行显著比较。不同萃取温度对萃取量的显著性比较见表4。

表1 不同料液比及萃取次数对萃取量的影响方差分析
注:*表示显著水平,**表示极显著水平。

表3 不同萃取温度、萃取时间及乙酸乙酯浓度对分离工艺的影响方差分析
注:*表示显著水平,**表示极显著水平。

表4 不同萃取温度、萃取次数、乙酸乙酯浓度对萃取量的显著性比较
结果显示在温度19~50 ℃之间,乙酸乙酯的萃取率随温度的增加而增大,四组数据均达到极显著水平。因此,选取萃取温度为50 ℃为最佳。
2.1.4 萃取时间对分离工艺的影响
称取3份雄烯二酮发酵废液5 g,按1∶3的料液比加入乙酸乙酯,分别在30、60、90、120 min的不同时间条件下,观察其分相情况与溶液的透明度,以萃取液上相溶液的高度为基准。不同萃取时间的分析比较见图1。
为判断不同萃取时间对分离甾醇工艺的影响是否显著,以确定最佳的萃取时间,对上述单因素进行方差分析,不同萃取时间对分离工艺的影响方差分析见表3。结果显示,不同萃取时间对萃取分离甾醇的结果未达到极显著水平,为了确定不同萃取时间与分离效果的影响差异,采用LSD(最少显著数法)进行显著性比较。不同萃取时间对工艺影响的显著性比较见表4。结果表明萃取时间在30~120 min范围内,30 min与120 min有显著性差异,60 min与90 min差异不显著,考虑对分离提取率的效果,选择萃取时间为120 min为佳。
2.1.5 萃取次数对分离工艺的影响
称取雄烯二酮发酵废液5 g,按料液比1∶3加入乙酸乙酯15 mL,分别按1、2、3次进行萃取,在恒温水浴50 ℃放置2 h后,上相离心后置于0 ℃条件下结晶6 h,离心称量沉淀物湿重,不同萃取次数的比较见表5。

表5 不同萃取次数的比较
为判断不同萃取次数对分离甾醇工艺的影响是否显著,以确定最佳的萃取次数,对上述单因素进行方差分析,不同萃取次数对分离工艺的影响方差分析见表1。结果显示,不同萃取次数对萃取分离甾醇的结果达到极显著水平,为了确定不同萃取次数与分离效果的影响差异,采用LSD(最少显著数法)进行显著性比较。不同萃取次数对工艺影响的显著性比较见表2。
结果分析:在萃取次数分别为1、2、3次萃取范围内,对甾醇的分离均达到极显著差异,根据分离的效果比较,选择萃取次数为两次最佳。
2.1.6 不同浓度乙酸乙酯对分离效果的影响比较
分别称取四份雄烯二酮发酵废液5 g,按料液比1∶3分别加浓度为100%、96%、93%、90%的有机溶剂进行萃取,在恒温水浴50 ℃下萃取2 h,吸取上相溶液在-10 ℃离心10 min,转速4 000 r/min,并记录上相溶液体积。经离心后,取上清液倒入试管中,在0 ℃下结晶6 h,再次离心,称量沉淀物湿重,乙酸乙酯的浓度对分离效果的比较见表6。

表6 乙酸乙酯的浓度对分离效果的比较
为判断不同乙酸乙酯浓度对分离甾醇工艺的影响是否显著,以确定最佳的乙酸乙酯浓度,对上述单因素进行方差分析,不同纯度乙酸乙酯对工艺的方差分析见表3。结果显示,不同乙酸乙酯浓度对萃取分离甾醇的结果达到显著水平,为了确定不同乙酸乙酯浓度对分离效果的影响差异,采用LSD(最少显著数法)进行显著性比较。不同浓度乙酸乙酯对工艺影响的显著性比较见表4。
结果分析:在乙酸乙酯不同浓度的范围中,纯度为100%、90%达到显著性差异,96%、93%未达到显著性差异,根据乙酸乙酯分离甾醇效果的比较,考虑溶剂的循环有效利用,选择乙酸乙酯浓度为90%为最佳条件。
2.2 冷却结晶法分离甾醇的工艺参数优化
2.2.1 不同结晶温度对工艺影响的单因素实验
分别称取3份雄烯二酮发酵废液5 g,萃取两次,在恒温水浴50 ℃下2 h后,经萃取后,取上相于-10 ℃离心10 min,转速4 000 r/min,取上清液倒入小试管中,并编号,在10、0、-10 ℃条件下冷却结晶6 h,观察其结晶情况与现象。然后过滤,称量沉淀物的湿重。不同结晶温度的分析比较见表7。
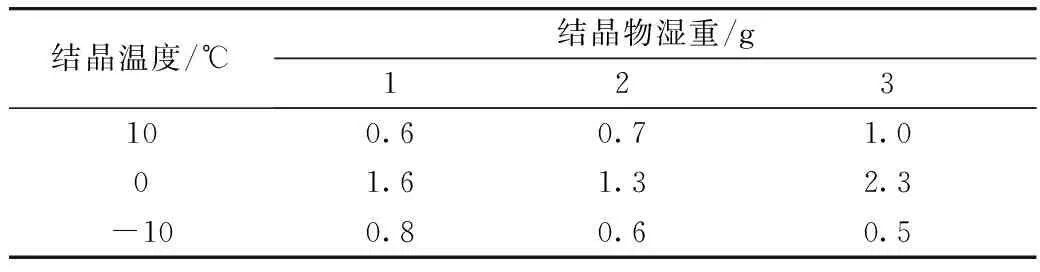
表7 不同结晶温度的分析比较
为判断不同结晶温度对分离甾醇工艺的影响是否显著,以确定最佳的结晶温度,对上述单因素进行方差分析,不同结晶温度的对分离工艺影响的方差分析见表8。
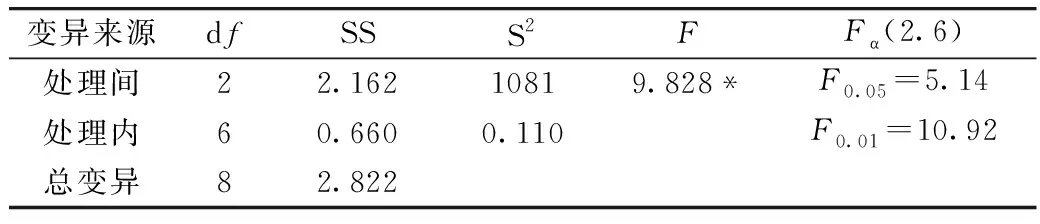
表8 不同结晶温度的对分离工艺影响的方差分析
注:*表示显著水平,**表示极显著水平。
结果显示,不同结晶温度对萃取分离甾醇的结果达到显著水平,为了确定不同结晶温度与分离效果的影响差异,采用LSD(最少显著数法)进行显著性比较。不同结晶温度对工艺影响的显著性比较见表9。

表9 不同结晶温度对工艺影响的显著性比较
结果分析:在0、10、-10 ℃之间,只有0 ℃与-10 ℃达到显著性差异,10 ℃未能达到显著性差异,根据对分离甾醇提取率的影响,选择0 ℃为最佳结晶温度。
2.2.2 不同结晶次数及结晶温度对工艺影响的两因素实验
分别称取3份雄烯二酮发酵废液5 g,按1∶3的料液比加入乙酸乙酯,在恒温50 ℃水浴中萃取2 h,并萃取两次,取萃取上相溶液于-10 ℃离心10 min,转速4 000 r/min,取上清液倒入小试管中,并编号,在0 ℃条件下,结晶6 h后,经过滤,沉淀物称量其湿重,而滤液再次倒入试管,放入冰箱结晶6 h,然后过滤,并再次称量其湿重,取滤液再次结晶6 h。再次过滤,称量沉淀物湿重。不同结晶次数及结晶温度的分析比较见表10。
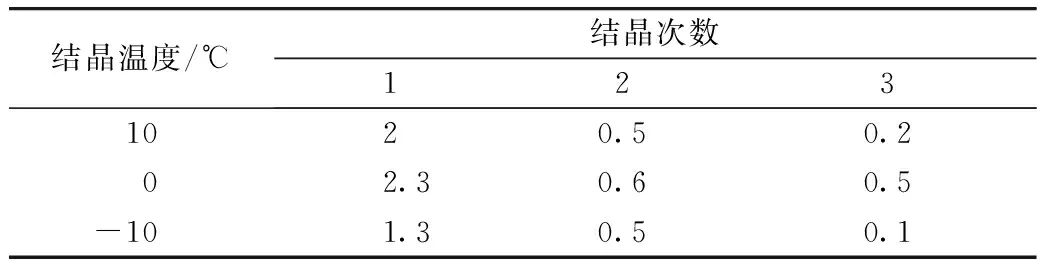
表10 不同结晶次数及结晶温度的分析比较
为判断不同结晶次对分离甾醇工艺的影响是否显著,以确定最佳的结晶次数,对上述单因素进行方差分析,不同结晶次数对分离工艺影响的方差分析见表11。
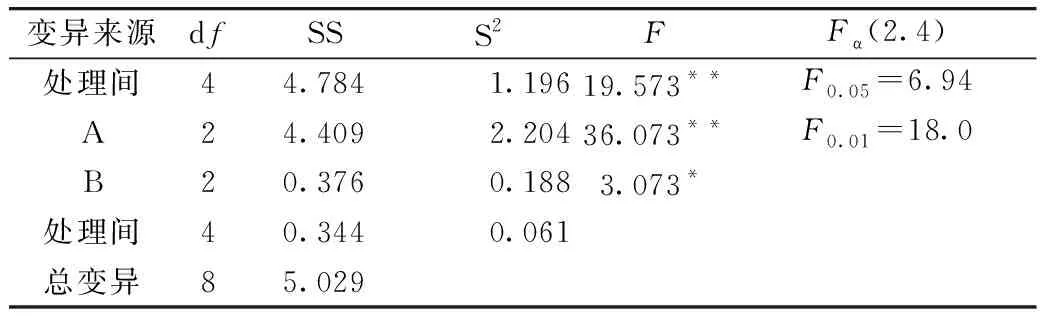
表11 不同结晶次数及结晶温度对分离工艺影响的方差分析
注:*表示显著水平,**表示极显著水平。
结果显示,不同结晶次数对萃取分离甾醇的结果均达到极显著水平,为了确定不同结晶温度和结晶次数对分离效果的影响差异,采用LSD(最少显著数法)进行显著性比较。不同结晶次数对工艺影响的显著性比较见表12。

表12 不同结晶次数对工艺影响的显著性比较
结果表明在不同的结晶次数下,1、2次均达到显著性差异,3次未能达到显著性差异,根据结晶次数对分离甾醇提取率的影响,选择最佳结晶次数为2次。
2.3 验证实验
按照上述工艺进行验证实验,以乙酸乙酯为溶剂,按料液比(1∶3)萃取温度50 ℃、萃取两次、结晶温度为0 ℃、结晶时间为6 h、结晶二次得到甾醇提取率为46%,纯度达到80%以上;按料液比1∶3加入甲醇溶解浓缩物,浓缩到体积的一半后,冷却结晶18h,离心,沉淀物得到含雄烯二酮粗品的产品,提取率为66.61%。上清液再次浓缩,回收溶剂和副产品大豆油。
3 结论
3.1 有机溶剂异丙醇、正己烷、石油醚对甾醇和雄烯二酮均有良好的溶解性,但在一定温度条件下分相效果上不佳,原因是发酵液中会形成乳化剂,使得分相难。实验选择采用中等极性的乙酸乙酯溶剂,在不需加任何助滤剂的情况下就可以自然沉降,从而解决工业上需要离心需消耗大量试剂的问题。
3.2 甾醇和雄烯二酮在大豆油中均具有良好的溶解,在发酵液中加入20%的大豆油时,甾醇的转化率达到到85%以上,在经过乙酸乙酯萃取后,分离甾醇后,可以浓缩回收溶剂以及大豆油,使得大豆都可以同时重复使用,与其他研究学者所研究的两相系统比较[2],工艺更加简便,在工业上更具有可行性,而且分离效果也较好。
3.3 通过单因素实验考察乙酸乙酯的倍数、萃取温度、萃取时间、萃取次数对分离甾醇的影响,确定最佳工艺条件为:萃取料液比1∶3,萃取温度50 ℃,萃取次数为二次,萃取时间为2 h。与文献报道酶法甲酯化提取甾醇相比较[12],该工艺操作更加简单可行。
猜你喜欢
山西化工(2022年4期)2022-09-23
现代临床医学(2021年5期)2021-11-02
昆明医科大学学报(2021年8期)2021-08-13
环境保护与循环经济(2021年12期)2021-03-16
食品安全导刊(2020年23期)2020-12-03
科学与财富(2020年26期)2020-11-16
食品安全导刊·中旬刊(2020年8期)2020-09-22
热带农业科学(2019年12期)2019-03-20
农民致富之友(2017年19期)2017-10-21
科学与财富(2017年17期)2017-06-16
