
HPLC法对黄精颗粒和提取物的色谱研究
2019-02-11 10:30孙艳平王荣杨宽申旭霁汪兴军
贵州医药 2019年12期
孙艳平 王荣 杨宽 申旭霁 汪兴军
(西安医学院药学院,陕西 西安710021)
黄精(P sibiricum Red.)是临床上重要的中药材,全球约有50多种,主要分布在北温带、北亚热带,而我国具有31种,多分布在南方热带以外地区[1]。《中国药典》2010版收载的黄精原植物有三种:黄精(P.sibiricum Red.)、多花黄精(P.cyrtonema Hua)、滇黄精(P.kingianum Coll.et Hemsl)。黄精的药用部位是根茎,能补气养阴、健脾、润肺、益肾作用[2]。用于脾胃虚弱,体倦乏力,口干食少,肺虚燥咳等症状。黄精身份相对较多,包括:木质素、多糖、氨基酸、皂苷类、微量元素等,且以多糖、皂苷类含量较高[3]。现代药理 结 果 表 明[4-5]:黄 精 具 有 抗 衰 老、调节机体免疫、改善记忆、抗肿瘤等作用。黄精颗粒属于常用的中成药,且与传统药物相比具有药物使用方便、安全性高等特点,但是临床上对于其药物成分缺乏统一的质量标准[6]。高效液相色谱法简称HPLC,是在经典液相色谱法的基础上,引入了气相色谱法的理论和实验技术,以高压输送流动相,采用高效固定相及高灵敏度检测器,发展而成的现代液相色谱分析方法[7]。本研究以黄精颗粒和提取物为对象开展研究,探讨HPLC 在黄精颗粒和提取物的含量测定效果。报道如下。
1 材料与方法
1.1 (1)仪器:托盘天平;分析天平;碾槽;量筒;烧杯;玻璃棒;纱布;蒸发皿;标准筛;砂锅;Agilent Technologies 1220Infinity LC高效液相色谱仪,购于美国安捷伦科技有限公司;Agilent HC-C18(2)色谱柱(4.6 mm×250 mm,5μm),KQ-250B 型超声清洗器,购于昆山市超声仪器有限公司;容量瓶;移液管;注射器;0.45μm 微孔滤膜;进样针。(2)试剂为黄精粉末;黄精颗粒;色谱乙腈(美国制造)(批号:20130318);薯蓣皂苷元标准品(中国药品生物制品检定所)(批号:201403181);纯净水。
1.2 黄精颗粒及提取物的制备 (1)黄精颗粒制备:取黄精药材100g于碾槽中碾碎取出,放入砂锅中加入800 mL 的自来水,浸泡20 分钟,煎煮30min,过滤除杂,滤渣加入400 mL 自来水,煎煮30min,过滤除杂,合并滤液,浓缩至浓度为1.1左右(90~100mL),换到水浴锅继续浓缩至玻璃棒挑起成丝,称量,记录浸膏的量,趁热加入蔗糖边加边搅至成团,慢慢加入糊精边加边制成握之成团,触之即散状态,过16目筛,过筛后的颗粒在60-80 ℃烘箱中干燥,整粒,备用[8]。(2)黄精颗粒提取物制备:分别精密称取黄精颗粒7g(相当于原药材2g)和药材粉末2g,置具塞锥形瓶中,按料液比为1∶20加入80%的乙醇,超声提取2次,温度60 ℃,时间50min,过滤,合并滤液,蒸干,加入20mL 蒸馏水溶解,加入20 mL 的正丁醇萃取二次,正丁醇层置于具塞锥形瓶中保存;向锥形瓶中加入少量甲醇溶解,分别置于10mL的容量瓶中,加入甲醇定容,置于2 ℃冰箱中保存,备用[9]。取样品提取液1mL,加5%α-萘酚乙醇液3滴,摇匀后沿试管壁缓缓加入浓硫酸,观察试管内液体颜色变化并记录。
1.3 方法
1.3.1 色谱条件 Agilent Technologies 1220Infinity LC 高效液相色谱仪,Agilent HC-C18(2)色谱柱(4.6 mm×250 mm,5μm),以乙腈(A)和水(B)为流动相,采用梯度洗脱,梯度程序为:0 min:5% A;10 min:10% A;30 min:35% A;40 min:40% A;60 min:5% A。流 速1 mL/min,柱 温30 ℃,检测波长200nm,进样量20μL[10]。
1.3.2 对照试验的配置 精确称取薯蓣皂苷元适量,加甲醇溶解配置成浓度为0.102 0mg/mL 的对照品溶液,0.45μm 的微孔滤膜过滤,备用。
1.3.3 高效液相图谱供试品溶液制备 分别将干燥至恒重的药材粉末2g和黄精颗粒精密称取7g(相当于原药材2g),置具塞锥形瓶中,按料液比为1:20加入80%的乙醇,超声提取2次,温度60℃,时间50min,过滤,合并滤液,蒸干,加入20mL 蒸馏水溶解,加入(20mL,20mL)的正丁醇萃取二次,取正丁醇层,旋干,加入少量甲醇溶解,分别置于10mL 的容量瓶中,加入甲醇定容,0.45μm 微孔滤膜过滤,取续滤液作为供试品。
1.3.4 精密度实验 取黄精颗粒1号连续进样5次进行测定,分别对共有峰保留时间和峰面积进行统计,计算RSD 值,结果发现它们的相对标准准偏差(RSD)均小于3%,因此,结果表明该仪器具有良好的精密度,见表1。

表1 精密度考察结果
1.3.5 稳定性实验 将配好的薯蓣皂苷元标准品按照1.4.2含量测定的色谱条件分别上样(8、10、12、14、16、18、20μL),以上样量和峰面积做标准曲线见表2和图1。 结果显示:薯蓣皂苷元在0.816μg~2.04μg内线性良好,回归方程:Y=20.662X+19.211,R2=0.999 4,R=0.999 7。
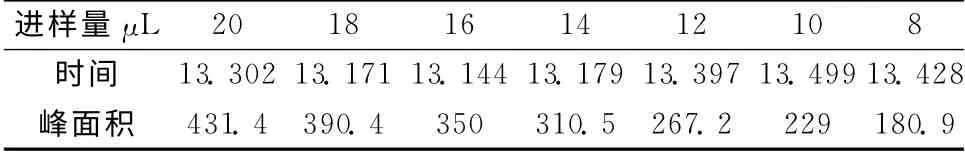
表2 薯蓣皂苷元的线性考察结果
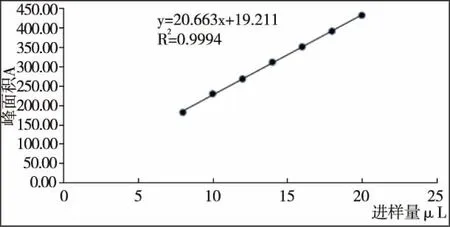
图1 薯蓣皂苷元的线性关系
1.4.6 重现性实验 取黄精颗粒3号供试品5份,按照1.3.2供试品溶液的制备方法和1.4.1色谱条件进行测定,统计各共有峰的保留时间和峰面积,计算RSD 值。结果如下图表明它们的相对标准偏差(RSD)均都小于3%,因此表明该仪器的重现性良好,见表3。

表3 重复性考察结果
1.4.7 稳定性实验 取黄精颗粒1 号供试品,在0、2、4、8、12小时依次进样测定,统计各共有峰的保留时间和峰面积,计算各共峰的RSD 值。结果表明它们的相对标准偏差(RSD)均小于3%,因此说明该仪器在12小时内的稳定性良好,见表4。

表4 稳定性考察结果
2 结 果
经过与标准品对比可以看书5号峰是薯蓣皂苷元。通过颗粒和原药材色谱图对比得知颗粒和原药材中的化学成分没变化,黄精颗粒中薯蓣皂苷元的含量比原药材中的薯蓣皂苷元含量高,见表5—7和图2—4。

表5 黄精颗粒各峰的峰面积和保留时间

表6 原药材各峰的峰面积和保留时间

表7 含量测定结果
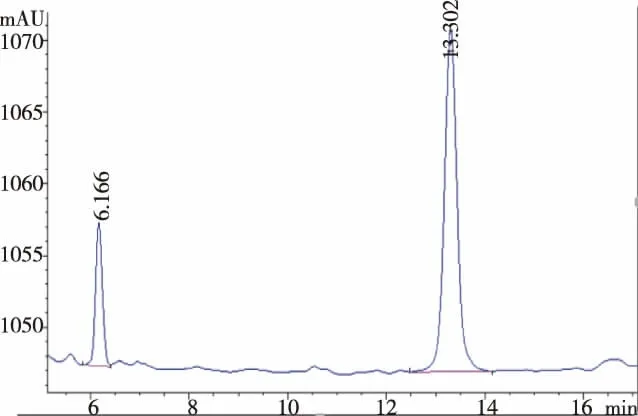
图2 薯蓣皂苷元标准品色谱
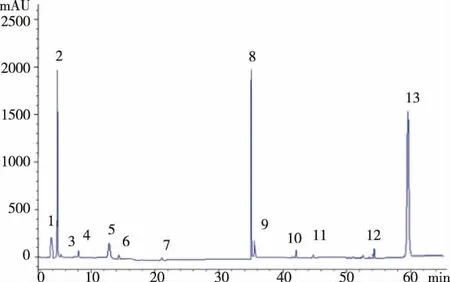
图3 黄精原药材色谱图
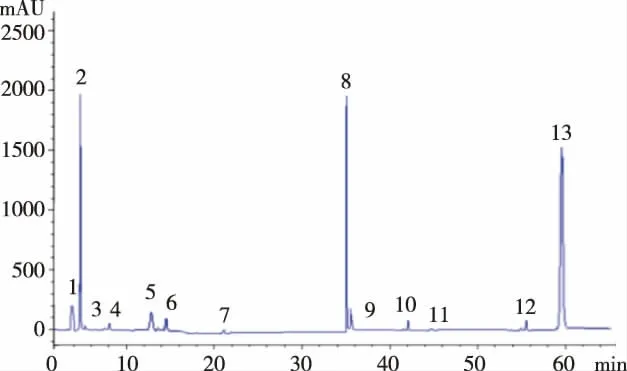
图4 黄精颗粒的色谱图
3 讨 论
黄精颗粒是临床上常用的中成药物,具有滋补肾精、益气补血功效,广泛用于月经量少、后错患者中[11-12]。但是,黄精颗粒临床制备难度较大,本研究中黄精颗粒制备过程中多次均无法制备出颗粒,并且制备的颗粒均难以制成团,难以完成药物过滤、筛选,经多次尝试后发现,制备颗粒时需要在药材浸润成膏时给予蔗糖作为敷料,边加边搅拌成稠状,然后在慢慢加入糊精制握之成团触之即散的状态[13]。同时,本研究中制剂、原料均给予超声法提取,但是由于原材料中杂质相对较多,抽滤时可给予提取液通过滤纸极慢。因此,本研究中给予棉花将原药材提取物中的杂质过滤一遍,然后再抽滤,这样使得提取液更容易通过滤纸。此外,HPLC 制备时以甲醇、乙腈、水为流动相选择最适合的流动相,通过实验发现流动相以乙腈-水为佳。然后以乙腈-水为流动相采用二元梯度洗脱。通过不断的更换流动相梯度,最后以1.4.1 高效液相图谱的色谱条件中的梯度扫出的峰相对较多,峰型相对较好。因此选用1.4.1高效液相图谱的色谱条件中的梯度进行实验。
查阅文献资料[14],选择薯蓣皂苷元的检测波长(200nm、210nm、203nm)进行测定,发现检测波长在200nm 处出峰较号而且出峰较多。因此选用检测波长为200nm 进行色谱研究。在方法学考察中对标准品线性进行考察时,刚开始扫出峰时间长而且杂峰多,还以为是标准品有问题,因此又换了不同厂家的标准品进行测定结果样品不仅不出峰而且高效液相仪的梯度泵压几乎不到1,经过分析才发现可能是高效液相色谱仪器工作时间太长了,因此关了仪器等了几天后使用仪器才恢复正常,标准品出峰的时间也比原来的出峰时间缩短了,而且峰性较好,峰面积稳定[15]。含量测定是制出的颗粒中薯蓣皂苷元的含量高于原药材中薯蓣皂苷元的含量,可能是由于实验存在正负误差或者是由于颗粒在提取的时候比原药材多提取了一次,因此做出这样的结果,结果表明:经过与标准品对比可以看书5号峰是薯蓣皂苷元。通过颗粒和原药材色谱图对比得知颗粒和原药材中的化学成分没变化,黄精颗粒中薯蓣皂苷元的含量比原药材中的薯蓣皂苷元含量高。
综上所述,将HPLC法用于黄精颗粒和提取物含量测定中效果理想,具有准确、简便,重现性好等优点;黄精颗粒中的薯蓣皂苷元含量比原药材中的薯蓣皂苷元含量高。
猜你喜欢
基层中医药(2022年2期)2022-07-22
Digital Chinese Medicine(2020年3期)2020-11-03
云南农业科技(2020年5期)2020-10-14
天然产物研究与开发(2018年8期)2018-09-10
中成药(2017年9期)2017-12-19
中成药(2017年9期)2017-12-19
医学研究杂志(2015年8期)2015-06-22
药学与临床研究(2015年4期)2015-06-05
特产研究(2014年4期)2014-04-10
饮食科学(2009年3期)2009-03-23
