
ATM缺失阻碍骨骼肌蛋白质合成的研究进展
2019-01-17 03:01冯祎中
浙江体育科学 2019年1期
金 晶,冯 燕,冯祎中
(1.浙江农林大学 体育军训部,浙江 杭州 311300;2.华东师范大学 体育与健康学院,上海 200241)
恶性体质(cachexia)是一种逐渐被认知的慢性病发生发展的疾病综合体,包括慢性心力衰竭、慢性肾病、慢性阻塞性肺部、骨骼肌衰减征等[1]。恶性体质不但引起财政和医疗的巨大负担,有较多的报告显示恶性体质与身体质量指数(body mass index,BMI)和骨骼肌丢失有高度相关性[2,3],且增加个人致残率甚至导致死亡[1]。Arthur[4]报告美国超过160 000恶性体质的住院案例,恶性体质人群住院时间中值为6天,平均消费超过10 000美元/人次,而非恶性体质住院者中值为3天,平均消费为4 000美元/人次。古希腊名医希波克拉底(公元前460-377年)描述心力衰竭病人写到:肉体被分解并开始化作水,腹部充水、腿脚肿胀,肩膀、锁骨、胸部和大腿被融化,这种病是致命的[5]。2009年德国诺贝尔文学奖获得者赫塔米勒同样冷淡地提及:当肉体逐渐从身体上消失,驮着骨架已成负担,驱使你埋进土地[6]。
毛细血管扩张共济失调(ataxia telangiectasia,AT)也是恶性体质的一种,案例研究报告AT病人的身材矮小生长停滞[7]、骨骼肌力下降[8],毛细血管扩张共济失调突变(ataxia telangiectasia mutated,ATM)与骨骼肌蛋白质合成有关[9]。尽管文献对ATM与mTOR通路的氧化应激[10]和DNA损伤[11]有所描述,但在ATM与蛋白质合成的分子机制描述并不全面,如Ching JK[9]只研究ATM对IGF-1/PI3K/Akt通路的影响,Burnett PE[12]只针对研究ATM对mTOR信号通路下游信号分子的影响,相关研究都提示ATM通过干预蛋白质合成呈现个体身材矮小、骨骼肌丢失等现象。本文拟通过对AT疾病和骨骼肌相关表型回顾,综述ATM与相关分子的调控关系,系统描绘ATM阻碍骨骼肌蛋白质合成的可能途径。
1 AT疾病和ATM
1.1 AT的症状
1994年统计美国和英国地区的发病率评估在1/40 000—1/100 000,AT杂合型女性拥有很高比例的乳腺癌风险,携带者的频率为0.5%~1.0%[13],在美国高加索人群中比例高达2%[14],现阶段还没有行之有效的治疗手段。针对美国地区出生儿童进行统计,报告发病率在1/40 000~1/300 000,并且疾病表型发生于幼儿时期,尤其在2~5岁已开始[15]。AT疾病是进程性的,随着年龄增长淋巴瘤和神经恶化进而增加死亡率和致残率,平均预期寿命在25岁[16,17]。
在AT基因被定以前,所有的文献都认为AT病人有极高的小脑共济失调和眼部及皮肤毛细血管扩张的发生率(100%的案例)[18,19]。然而,在AT基因被定义之后,有很多文献发现AT基因的病例没有发生小脑共济失调或眼部的毛细血管扩张:Trimis[20]等报告6岁女孩遗传学上被确认为AT病人,但是没有发生神经系统的病变;Alterman[21]等研究两兄妹发现虽然有严重的细胞表型,但只存在轻微的神经临床表型;moin[22]等评定临床和实验室的104名AT病人,发现所有病人都存在小脑的共济失调,而眼球和眼部皮肤的毛细血管分别发生87和73人。
1.2 ATM的发现与定义
毛细血管扩张共济失调突变(ataxia telangiectasia mutated,ATM)是常染色体上隐性基因不稳定发生突变形成以小脑共济失调、免疫缺失、癌症和呼吸系统易感染等特征的综合症[23],称为“A-T”(ataxia telangiectasia,毛细血管扩张共济失调)。AT还叫作路易斯-巴尔二氏综合症,也经常用艾琳娜博德和罗伯特塞奇威克,第一次被描述为家族性的退行性小脑失调症状[24]。ATM基因位点最早是由Gatti[25]等通过数理分析共分离统计定位于第11号染色体的q22.3-23.1,在随后的七年,许多研究所和科研工作者不断完善,最终由Savitsky[26]团队将其定义完成,它由66个外显子(4个非编码和62个编码)跨越150kb的DNA组成。ATM是~350kDa的高分子蛋白激酶,属于磷脂酰肌醇-3激酶相关蛋白激酶(phosphoinositidyl 3-kinase-related protein kinase,PIKK)家族成员[27],认为它在调控细胞循环和DNA损伤方面有重要作用[28,29]。
2 AT与生长、骨骼肌和运动能力
2.1 AT与身高、体重
绝大多数AT患者都发现有生长停滞和体重下降等特征[7,8,30-33]。研究报道AT病人的身高、体重以及身体质量指数(body mass index,BMI)均显著低于对照组[32,34]。Ehlayel[35]和Pommerening[8]等分别报告13名和25名AT病人与对照组比较,发现身高存在极其显著性差异。澳大利亚地区的13名AT病人(年龄4~23岁,其中女性9人)结果显示77%病人体型偏小(z score<-1),且54%的病人体重不足[36]。Pommerening[8]等研究发现AT病人的体重和BMI极其显著低于对照组,Schubert R[33]将19名AT患者(男性11人和女性8人,年龄为4~24岁)按年龄分为三组,A组≤12岁、B组12~18岁和C组>18岁。结果显示A组的BMI为15.3 kg/m2,B组的病人显示体重和生长发育困难引起BMI下降到正常范围内的3%之前,C组出现进一步下降的恶性变化,BMI尽然为12.7 kg/m2。与此结果相一致的研究还有英国地区70名AT病人的统计研究,54/70的病人出现极其偏瘦[30];Kieslich M[31]团队对11名病人(8~26岁)进行体重和BMI的分析结果均显示是显著低于对照组(P<0.05);Schubert R[33]对19名AT病人尽管给予充足的营养条件,BMI仍然显著下降。
另外,AT的动物模型通过敲除ATM基因即ATM-/-小鼠个体,同样也出现生长延缓或停滞现象[37,38]。James KC团队发现ATM敲除小鼠纯合子具有小体型,而杂合子ATM+/-与野生型小鼠体重没有差异性,而ATM-/-小鼠的体重是野生型的70%,与AT病人体重下降结论相一致[9]。
2.2 AT与骨骼肌及运动能力
骨骼肌是人体构造的重要组成部分,是氨基酸合成蛋白质的储水库[39]。厌食、脱水、恶性体质和骨骼肌衰减征等引起骨骼肌丢失最常见的原因,造成肌肉进程性的下降病理生理学的原因是复杂和多因素的,比如久坐不运动、疾病、衰老和营养状况等都有因果联系[40]。近期报告提示ATM与骨骼肌丢失存在很大的相关性,发生骨骼肌的丢失后进而直接或间接影响个体的运动能力[7,8,34]。
2.2.1 AT与骨骼肌。研究报告许多疾病所构成的恶性体质(cachexia)都会造成骨骼肌质量的大量丢失,进而呈现个体运动能力受限和功能状态的低下[1]。例如心力衰竭病人的临床表现为心肌衰竭,限制其运动能力,骨骼肌质量和力量下降从而影响其运动能力[2];Martin L[3]等研究证明所有癌症病人均出现体重下降现象,肌肉指数下降和肌肉衰减的症状;AT患病个体无论是个案研究还是群体调查都发现体重和骨骼肌力下降及肌肉衰减等症状[7,8]。
Da silva R[41]在研究AT病人时发现有很高比例患者出现降低瘦体重(Fat Free-mass,FFM)现象,Pommerening H[8]同样也发现FFM在AT人群中含量显著低于对照组,并且认为AT病人的脂类和脂肪组织没有发生变化,也就是说体重下降主要是减少骨骼肌质量。Heil JA报告4名来自两个家庭的成年AT病人,患有脊椎肌肉萎缩症并伴有股四头肌的萎缩;Dunn HG[42]针对加拿大AT病人进行尸检,发现骨骼肌出现轻微的萎缩现象。利用液压测力计测定AT病人的力量显著低于对照组[8],Felix E[32]发现病人的初始呼吸肌的力量和肺活量显著低于对照组,经过24周的呼吸肌训练,有效的加强肺活量和呼吸肌力量,极大改善AT病人的生活质量。
2.2.2 AT与运动能力。英国70人案例研究发现,43/68大部分患者报告3岁之前发生运动失调症状[30],另一项最新的个案研究显示病人从34周龄出生、6月坐、11月站立、14月行走都是正常的,而从两岁开始行走能力发生改变,需要外力的支撑、身体开始呈现不稳定状态及频繁摔倒等现象出现[7],与gatti RA[43]报告运动失调发生在2~3岁吻合。Teive HA[14]等在研究AT的综述中表述,5~10岁出现更为严重的神经性病变导致运动能力的缺失、徐动症的发生。Felipe RB[7]等报告的个案在6岁时出现进阶式的运动失调和颤抖,7岁时完全依靠外界控制站立,四肢末梢开始颤抖,只能依靠某一姿势控制身体;较早的文献也报告AT病人手臂出现颤抖现象而且精准度下降,行走困难有摇晃状态,站立和坐姿都显示发躯干弯曲等现象[42]。Woods CG[30]统计研究认为病人在8岁失去书写能力,平均10岁左右失去行走能力。年龄对于AT病人的行走能力有很大相关性,行走组(gait preserved)和轮椅组(wheelchair-bound)之间年龄存在显著差异;AT病人的去脂体重和体细胞质量极其显著低于对照组;维生素D的含量AT病人显著低于对照组,有趣的发现所有缺乏维生素D的病人都是大于12岁并且失去行走能力全部为轮椅组的[8]。
3 抗阻运动诱导骨骼肌肥大及其合成机制
3.1 抗阻运动诱导骨骼肌肥大及IGF-1的重新分配
抗阻运动是肌肉受到剧烈收缩进而刺激骨骼肌肥大的有效方式[44,45]。Luciano TF[44]构建3种不同的抗阻模型对比各方案中大鼠骨骼肌横截面积、质量和蛋白质合成等,结果显示所有抗阻模型经过训练后骨骼肌的体积得到显著提升;Tang JE[46]招募普通青年男子通过12周的抗阻训练后,骨骼肌活检前后比较,发现骨骼肌的横截面积显著增加;Salvadego D[47]则通过长期从事健美的运动员与对照组进行对比,结果显示股四头肌的横截面积和体积显著大于对照组(P<0.05);Ogasawara[45]等采用动物模型通过电刺激模拟抗阻运动,研究发现腓肠肌的净含量和相对体重比值,单次急性刺激后均没有变化,而通过12次和18次训练后分别增加8.6%和10.7%。
普遍认为抗阻运动是通过IGF-1/PI3K/Akt通路激活mTOR,继而促进骨骼肌的蛋白质合成[48]。Arnarson A[49]观察全身骨骼肌中胰岛素样生长因子(insulin-like growth factor-1,IGF-1)的分布情况,发现IGF-1经过12周抗阻运动干预后有很强的变化,下调血清中总IGF-1的含量。血清IGF1经过抗阻运动后实验报告下调或不变:Mangine[50]对比高强度与中等强度抗阻运动,研究结果显示高强度组的血清IGF-1显著下调;另外的研究认为抗阻运动后未造成血清IGF-1的改变[51,52]。相对于血清的变化,骨骼肌中的IGF-1显著上调,Kido[51]实验通过急性抗阻运动后,测试1h和3h后骨骼肌的IGF-1发现显著上调;相似的另一项研究检测骨骼肌IGF-1浓度在1h和6h后上调[52]。抗阻运动干预实验,总体血清IGF-1始终保持不变或下调,而骨骼肌中IGF-1上调,推测抗阻运动诱导骨骼肌中IGF-1在全身的重新分配,特别是骨骼肌中高表达的IGF-1参与骨骼肌蛋白质合成过程[49,50]。
3.2 骨骼肌蛋白质合成经典机制
运动诱导骨骼肌生理性适应肥大机制涉及到多种信号传导途径,其中雷帕霉素靶体蛋白(mammalian target of rapamycin,mTOR)信号传导通路目前备受关注[53]。mTOR是非典型丝氨酸/苏氨酸蛋白激酶,在进化上高度保守,其分子量大小是289kDa,是磷脂酰肌醇激酶相关激酶PIKK蛋白家族成员[54]。mTOR具有广泛的生物学功能,可整合营养、能量及生长因子等多种细胞外信号,参与基因转录、蛋白质翻译、核糖体合成等过程,在调控蛋白质合成、细胞增殖、凋亡、自噬等细胞生长过程中发挥及其重要的作用[54]。
大量的文献证明mTOR在调控蛋白质合成和骨骼肌细胞增长/肥大过程中起到关键作用[53,55,56]。以mTOR为关键节点本文将mTOR上游信号通路(IGF-1/PI3K/Akt通路和TSC1/2通路)和下游信号通路(4E-BP1/eIF4E通路和S6K1通路)分别进行详述。
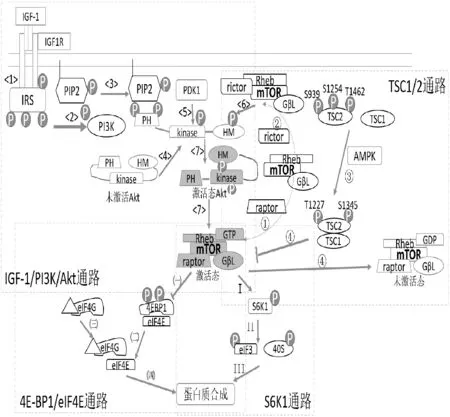
图1 导致骨骼肌细胞肥大的主要胞内信号传导途径(综合参考文献[57-59]中的各图重新绘制)
3.2.1 IGF-1/PI3K/Akt通路。在IGF-1介导的通路中发生信号级联放大,这种放大反应起初发生在细胞膜上,由IGF-1与IGF-1受体(insulin-like growth factor-1 receptor,IGF-1R)相结合,可引起IGF1R的磷酸化。IGF1R自身磷酸化后,激活细胞内酪氨酸激酶进而促使下一靶点蛋白胰岛素受体底物(IRS)的磷酸化[48,60](图1,途径<1>)。该反应引起IRS的大量募集,IRS是IGF1R和PI3K(磷脂酰肌醇-3-激酶)之间的衔接蛋白,其磷酸化能进一步激活PI3K[9](图1,途径<2>)。被激活的PI3K可以催化位于细胞膜磷脂双分子层内层上的PI(4,5)P2肌醇环3位上继续添加一个磷酸基团形成PI(3,4,5)P3(图1,途径<3>)。骨骼肌细胞膜的内膜上一旦有PIP3的形成,呈未激活态的Akt就会大量被募集到胞浆内膜上,并使其被激活[61]。Akt又称为PKB(蛋白激酶B)是一类丝氨酸/苏氨酸蛋白激酶,完整的蛋白质由三大部分所构成:氨基端的pleckstrin homology(简称PH)区、中间的激酶(kinase)区和羧基端的疏水作用(简称HM)区。在未激活态/原始态的三级构象中,Akt的PH区和HM区距离相对较近,当其PH区结合到膜内层的PIP3上后,整个蛋白质分子呈现平铺拉伸构象,促使激酶区与HM区得到充分暴露(图1,途径<4>)。随后,Akt上两个氨基酸位点:激酶区上的苏氨酸308(图1,途径<5>)和羧基端的HM区上的丝氨酸473均被磷酸化(图1,途径<6>),从而使Akt完全被激活,最终通过IGF-1/PI3K/Akt通路使mTOR进行磷酸化促其活性被激活[48](图1,途径<7>)。
3.2.2 TSC1/2通路。在哺乳动物细胞中mTOR可以分为两种复合物mTORC1和mTORC2。两种复合物之间的区别在于组成的亚结构不同,mTORC1包含raptor(regulatory associated protein of mTOR)、GβL(G-proteinβ-subunit-like protein)、Rheb(ras homolog enriched in brain)和mTOR(图1,途径①),而mTORC2含有rictor(rapamycin-insensitive companion of mTOR)和其他组成(图1,途径②)。两者都有特定而且唯一的下游靶蛋白,如mTORC1只对4E-BP1和S6K有磷酸化作用,而mTORC2只磷酸化蛋白激酶Cα和Akt[62]。Raptor结合于4E-BP1和S6K的区域作为mTOR信号基序,募集mTORC1复合物,通过mTOR进行磷酸化作用。因此可以认为raptor是紧密连接mTOR与其底物的衔接分子[57]。Rheb是GTP的激酶(GTPase),其结合GDP或GTP决定mTOR是否被激活,与GTP结合时对mTOR进行激活,与之相反,形成Rheb-GDP复合物时则抑制mTOR活性。mTOR复合体与GDP/GTP结合取决于GTPase 激活蛋白(GAP)tuberin(又称为TSC2)和另一种复合蛋白hamartin(TSC1)[63](图1,途径③)。研究发现,在AMPK的参与下TSC1和TSC2形成复合体TSC1/2可调控Rheb与GTP结合向与GDP结合方向转化,促使Rheb与GDP结合从而使mTOR活性抑制(图1,途径④);而磷酸化的Akt可以抑制TSC1/2复合体的活性,进而向Rheb与GTP结合的方向转变从而激活mTOR,增强其信号通路[57]。
3.2.3 4EBP1/eIF4E通路。mTOR的促合成通路依赖于下游信号4EBP1磷酸化后调节eIF4E的活性,用来提高翻译效率,进而促进翻译的起始过程和延伸过程,增加骨骼肌蛋白合成[64](图2)。
eIF4E是翻译启始因子,也是mTOR下游通路的重要蛋白,mRNA翻译的启始阶段是整个翻译过程的关键部分。活化的mTOR蛋白中含有的raptor结构可以直接/间接的方式结合TOR信号基序的4EBP1的C端,使其4EBP1磷酸化[65]。这个过程中eIF4E首先在5’mRNA末端与m7GTP的cap结构结合,eIF4E还与其他多种亚结构结合(如eIF4A和eIF4G)形成复合体,进而与40S核糖体结合发挥其作用[65]。4EBP1和eIF4G均能与eIF4E相结合而且反应发生是在相同部位,两者竞争同一底物即eIF4E是它们共同的结合靶点。mTOR至少可以磷酸化4EBP1的Thr37和Thr46两个位点[57],4EBP1通过磷酸化水平的高低来调控与eIF4E的结合情况,当4EBP1不被mTOR磷酸化时,低磷酸化状态的4EBP1与eIF4E具有较高的亲和力,4EBP1就与eIF4E结合;而当4EBP1被mTOR磷酸化后抑制它们结合(图1,途径㈠),处于较高磷酸化状态时降低了相互之间的亲和力则释放出eIF4E[59](图1,途径㈡),eIF4G则抢占作用靶点与eIF4E结合(图1,途径㈢)。eIF4E和eIF4G结合并与其他蛋白共同形成复合体和40S共同编码mRNA,最终促进骨骼肌中蛋白质的合成[58](图1,途径㈣)。
3.2.4 S6K1通路。mTOR的促合成效应除了4EBP1/eIF4E通路以外还需要通过S6K1磷酸化来实现。mTOR的下游信号S6K1磷酸化后,能促使5’TOPmRNA翻译,提高mRNA的翻译能力[66](图2)。
整个启动复合物结构非常复杂,主要由3个核心部分组成(图2):①eIF4E,结合于m7GTP(7-甲基-GTP)5’覆盖的mRNA,促进复合物与核糖体一起装配;②分子脚手架eIF4G,在启动子和40S核糖体亚结构之上的元件;③ATP依赖的RNA解旋酶eIF4A[67]。4E-BP1和S6K1是mTOR共同的磷酸化靶点,通过对下游的调控结合完成蛋白质合成。
通常情况下S6K1是附着在eIF3复合物上的,与mTOR/raptor共同结合于复合物上。在生长因子、营养物质等条件的作用下,mTOR激活后会使得S6K1上的Thr389磷酸化(图1,途径Ⅰ),磷酸化的S6K1随即就与eIF3复合物脱离。PDK1虽然不是复合物的一部分,但是它能与脱离的Thr389磷酸化的S6K1相互反应,催化作用发生下再次磷酸化S6K1的Thr229[59]。激活态的S6K1进一步磷酸化40S和eIF4B,一方面S6K1直接磷酸化40S核糖体蛋白S6[65];另一方面磷酸化后的eIF4B被募集到eIF3(图1,途径II),最终形成复合体发挥编码mRNA的作用[59](图1,途径III)。
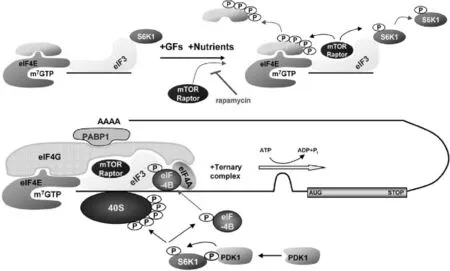
图2 mTOR下游特色效应器(4E-BP1和S6K1)的反应机制[59]
4 ATM影响蛋白质合成的相关分子机制
4.1 AT病人与IGF-1
在研究AT病人的血清指标发现大部分个体的胰岛素样生长因子-1(Insulin-like growth factor-1,IGF-1)含量普遍偏低,认为IGF-1及其相关蛋白可能是影响生长状况和体重下降的原因[31, 33]。Schubert R[33]测得9/16病人的IGF-1含量低于同龄对照的前3%,而且13/16病人的IGFBP-3(它是IGF-1主要结合蛋白)浓度显著较低。Voss S[68]对24名患者进行IGF-1的测定,发现其中10位(41.7%)的浓度低于同龄前3%,Kieslich M[31]也报告6名病人的含量低于前3%。Pommerening H[8]对激素指标进行测定,结果显示IGF-1、皮质醇和脱氢表雄酮显著低于对照组。从AT病人获得的成纤维细胞进行培养,IGF-1-sCLU(secretory clusterin)蛋白水平显著低于对照组,推测是由于IGF-1含量偏低造成这种现象[69]。
4.2 ATM与胰岛素/IGF-1刺激下的Akt磷酸化
Akt和ATM基因敲除模型小鼠显示如生长停滞、不育症、免疫系统缺陷和胰岛素抵抗等相似的表型[37,70]。一方面,磷酸化的Akt通过IGF-1/PI3K/Akt通路使mTOR进行磷酸化促其活性被激活[48],另一方面,磷酸化的Akt可以抑制TSC1/2复合体的活性,进而促进Rheb与GTP结合的方向转变从而激活mTOR[57],因此Akt的激活对于骨骼肌蛋白质合成至关重要。
ATM蛋白含量与细胞中Akt磷酸化水平在正常情况下并没有相关性,当受到胰岛素/IGF-1等条件的刺激下ATM的缺失直接影响Akt磷酸化[9,71,72]。Ching JK[9]利用10nM的IGF-1对比目鱼肌细胞和C2C12肌管进行20min的刺激,发现ATM KD的C2C12肌管显著下调Akt的S473/T308磷酸化水平;对比于ATM+/+小鼠的比目鱼肌细胞,ATM+/-的Akt S473/T308磷酸化水平发生明显的阻碍作用,进一步检测p-mTOR蛋白含量也观测到其显著下调,而总体mTOR没有变化;Ching JK[72]的另一项研究利用2mU/ml的胰岛素30min的刺激下,比目鱼肌中(ATM+/+和ATM-/-)两种基因型之间Akt的磷酸化没有差异性,而胫骨前肌的纯合型阻碍Akt两位点S473/T308磷酸化相较于野生型(P<0.05)。同样的Jeong[71]利用C2C12 ATM shRNA在100nM胰岛素的刺激下,直接阻碍Akt的磷酸化;Hresko RC[48]和Viniegra JG[73]对Cos细胞的ATM+/+和ATM KD进行分析,发现Akt S473的磷酸化在野生型中表达明显升高,AT病人(GM08931)和KO小鼠获得的细胞系在胰岛素的刺激下抑制Akt的S473磷酸化作用[73];小鼠的胚胎成纤维细胞模型ATM+/+和ATM-/-,也证明了ATM的缺失对Akt S473的磷酸化作用[74]。
KU55933是ATM的抑制剂,阻碍胰岛素刺激的Akt的磷酸化。Halaby MJ利用KU55933抑制小鼠L6肌细胞,抑制Akt的磷酸化[74];Jeong利用1μM的KU55933分别对L6肌管、C2C12、比目鱼肌和RD(分化后的横纹肌肉瘤)细胞进行干预,发现C2C12和RD细胞中抑制ATM使得Akt的S473磷酸化有明显的下调作用,L6肌管细胞在1μM KU55933+100nm/25nm的胰岛素都没有对Akt磷酸化有影响[71]。过量表达的ATM促进野生型诱导S473的磷酸化,但在ATM KD型中没有变化,Akt的T308磷酸化也没有差异性;另一种方式通过siRNA技术外源性感染细胞抑制ATM的表达,结果显示显著下调S473的磷酸化[73]。综上所述,ATM被抑制/缺失则下调Akt的磷酸化,而当过量表达时上调其磷酸化水平,因此认为ATM在胰岛素/IGF-1的刺激下正调控Akt的磷酸化水平,特别是S473磷酸化水平。
4.3 ATM与Akt的上游信号通路
4.3.1 ATM与IGF-1R。IGF-1R是IGF-1的受体,是酪氨酸激酶的跨膜受体几乎所有组织中都表达,调控细胞的生长、分化、转化和凋亡有重要作用[75]。IGF-1R基因缺失也会造成体重下降,IGF-1R-/-小鼠的体量与野生型比较下降45%,出生后的模型鼠多个器官异常并导致死亡,另外IGF-1R-/-的小鼠胚胎时期普遍发生器官和骨骼肌的发育不良[76];另外两项细胞实验都认为ATM对IGF-1/IGF-1R有影响,Luo X[69]从AT病人获得的成纤维细胞进行细胞培养,发现突变细胞的IGF-1-sCLU(secretory clusterin)水平显著低于对照组,认为ATM缺失诱导IGF-1含量偏低造成这种现象,Goetz EM[77]将正常细胞暴露于DNA损害元件时发现提升IGF-1的表达量,进而激活IGF-1R通路和sCLU,然而ATM缺失的细胞不能诱导IGF-1R的表达。
Peretz S[78]在研究AT细胞时发现与杂合型/野生型相比,IGF-1R处于较低水平(P<0.05)。为了探寻ATM与IGF-1R的直接关系,转染完整ATM全片段的cDNA到AT突变细胞中[79],8个细胞GM5849通过实验技术Western blot验证cDNA全部成功转染,证明有完整表达的ATM cDNA能提高IGF-1R的表达量[78]。紧接着,在AT细胞中发现IGF-1R启动子活性下降,为了证明转染ATM cDNA的细胞中IGF-1R启动子区域活性也增强,将该区域用荧光素酶受体转染[80],结果显示转染后的ATM cDNA结合细胞中启动子的活性是ATM突变细胞的4倍;另一个实验利用ATM突变(GM3487)和父母杂合型(GM3489)获得的成纤维细胞进行启动子区域的荧光素酶转染,结果也证实了ATM+/-的启动子活性表达量显著高于ATM-/-[78]。Peretz S[78]利用转载体的转染到野生型成纤维细胞系中,与ATM蛋白的亮氨酸拉链结构相结合,抑制ATM蛋白的产生,结果证明ATM受到抑制后下调IGF-1R的表达量。
4.3.2 ATM与IRS-1和PI3K。IRS-1是IGF-1R的靶点,可作为IGF-1R和PI3K的衔接蛋白。从同一样本的不同位点中的p-IRS Y612和总IRS分别进行检测,结果显示C2C12细胞的野生型和杂合型在添加IGF-1刺激情况下p-IRS和总IRS-1都有显著性差异提高,胰岛素刺激下P-IRS-1/IRS-1的比值在杂合型中表达偏低,未达到统计意义[9]。胰岛素信号通路会被氨基末端激酶(Jun N-terminal kinase,JNK)诱导的IRS-1 S307的磷酸化所阻断,在ATM-/-动脉细胞发现JNK/总JNK和IRS-1 S307磷酸化/总IRS-1显著高于ATM+/-和ATM+/+,ATM缺失小鼠的脂肪组织、骨骼肌细胞以及肝脏都存在高活性的JNK,激活IRS-1 S307磷酸化阻碍胰岛素信号通路[81]。
Ching JK[9]利用激酶反应试剂盒检测比目鱼肌中野生型和杂合型的PI3K活性,结果显示IGF-1刺激下的PI3K活性在ATM+/-中偏低(P=0.053)。Viniegra JG[73]研究ATM对Cos细胞胰岛素刺激的Akt S473的磷酸化有直接作用,进一步探究是否通过抑制PI3K能够阻碍Akt的磷酸化,与预期的相一致,当加入PI3K的各种抑制剂(200nM渥曼青霉素、2μMLY294002、2nM咖啡因)完全阻碍S473的磷酸化,也就是说ATM-PI3K-Akt通路中通过抑制PI3K,直接阻碍Akt的磷酸化进行。
4.4 ATM与Akt的下游信号分子S6K1和4E-BP1
在活体雷帕霉素影响的研究中编码翻译过程受到许多磷酸化所调控,如核糖体S6蛋白和其激酶p70S6K1以及eIF-4F结合蛋白4E-BP1。Ching JK进行3个单独实验验证ATM在IGF-1刺激下促使S6K1磷酸化下调:①IGF-1刺激的S6K1磷酸化位点在T389,C2C12细胞ATM shRNA中S6K T389磷酸化水平显著下降;②正常的C2C12细胞(ATM+/+)培育在KU55933中,通过阻碍胰岛素通路中的PI3K,进而抑制通路进程来降低S6K1的磷酸化水平;③在观察只有一半功能的杂合型ATM+/-比目鱼肌细胞,在IGF-1刺激下S6K1磷酸化水平显著低于野生型对照[9]。Burnett PE[12]报告ATM类似家族的相关激酶有助于促进mTOR下游S6K1和4E-BP1磷酸化。Yang DG[82]在研究胰岛素刺激下蛋白质合成通路过程中,发现诱导eIF4E的结合蛋白4EBP1,ATM能在4EBP1的S111位点发生磷酸化,当ATM缺失的离体细胞,显著下调4EBP1从eIF4E分离水平。与此结果相一致的研究Kuang X[83]在ATM-/-小鼠中发现ATM的功能缺失下调mTOR信号通路的4EBP1水平。
5 ATM阻碍蛋白质合成的路径假设
AT疾病同样被归类于恶性体质,会呈现出骨骼肌合成受阻和肌力下降等典型表征[3],虽然AT疾病在世界范围内的发病率处于1/44 000—1/100 000之间并不高[14],但是自1964年Dunn首次发现至今[42],科学界对其仍然给予极大的热情[7]。与其他恶性体质的疾病不同,AT疾病是缺少单个蛋白(ATM)的表达[25],导致整体骨骼肌丢失和运动能力等表型下降,因此探索AT疾病与骨骼肌蛋白质合成的关系从实验的角度来看,就大大提高可行性和可信度。
宏观视域下,AT疾病与骨骼肌的关系在正文部分较为详细的论述,AT患者的身高、体重和BMI显著低于对照组,骨骼肌的体积、横截面和肌力显著下降,并且随年龄增长而渐进式加剧骨骼肌各项表征下降进而影响行走、站立等运动能力[8, 15, 35]。微观视域下,骨骼肌蛋白质mTOR通路是蛋白质合成的主要信号通路,本文梳理ATM与蛋白质合成mTOR通路中的关键分子的相关性,而且所有的结果一致认为ATM阻碍相关分子的蛋白质合成(即负向调控合成)[9],基本构建出ATM阻碍/抑制骨骼肌蛋白质合成的可能路径(见图3)。ATM缺失/下调可能发生的假设路径:下调ATM→下调IGF-1→下调IGF-1R→下调IRS-1(通过下调Y612或上调S307)→下调PI3K→下调Akt(通过下调S473和T308)→下调mTOR→下调S6K1和下调4E-BP1的分离率→骨骼肌蛋白质合成。
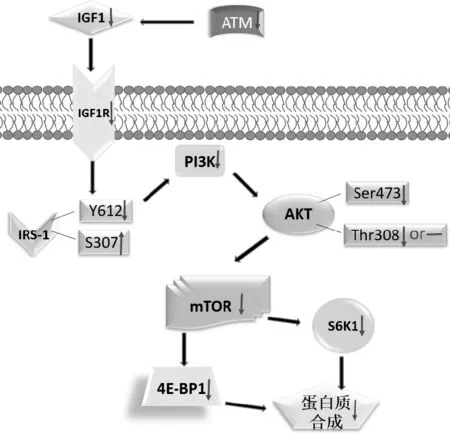
图3 ATM阻碍骨骼肌蛋白质合成的路径假设
不同肌纤维对胰岛素刺激下的ATM的作用有显著性不同(胫骨前肌的Ser473磷酸化,而比目鱼肌未发生)[72],而且还发现胫骨前肌的ATM蛋白水平是比目鱼肌中的10倍,表明ATM更倾向于快肌(胫骨前肌vs比目鱼肌)[9]。抗阻运动又是提升骨骼肌中IGF-1的方式,有氧运动并未发现能促进IGF-1的分泌[51],另一项研究证明AT病人经过呼吸肌的锻炼,力量得到显著提升[32]。抗阻训练对ATM蛋白缺失的个体有正向调控,并且提示可作为改善AT疾病病况和提升生活质量的非药物手段,因此,需要有更多的动物活体实验通过抗阻运动的干预,探索干预后AT疾病表征和骨骼肌蛋白质合成中ATM蛋白的有益作用。
猜你喜欢
体育科技文献通报(2022年3期)2022-05-23
波谱学杂志(2022年1期)2022-03-15
医学综述(2021年16期)2021-12-01
天津医科大学学报(2021年3期)2021-07-21
世界科学技术-中医药现代化(2021年12期)2021-04-19
天津医科大学学报(2019年6期)2019-08-13
天然产物研究与开发(2018年2期)2018-04-04
分析化学(2017年12期)2017-12-25
中国医药生物技术(2015年4期)2015-12-26
安徽医科大学学报(2015年9期)2015-12-16
