
利用WGCNA进行玉米花期基因共表达模块鉴定
2019-01-17 01:25杨宇昕桑志勤代文双
作物学报 2019年2期
杨宇昕 桑志勤,2 许 诚 代文双 邹 枨,*
利用WGCNA进行玉米花期基因共表达模块鉴定
杨宇昕1桑志勤1,2许 诚1代文双1邹 枨1,*
1中国农业科学院作物科学研究所, 北京 100081;2新疆农垦科学院, 新疆石河子 832000
权重基因共表达网络分析(weighted gene co-expression network analysis, WGCNA)是系统生物学的一种研究方法, 在挖掘生物学数据与特定性状之间的生物学关系方面具有十分重要的作用。本研究利用玉米(L.)自交系B73的14份不同发育阶段的转录组数据, 筛选掉低表达丰度的基因, 最终得到了22,426个高表达的基因用于创建基因表达矩阵; 利用不同组织作为性状, 创建表型矩阵。然后利用R软件中的WGCNA包建立了共表达网络, 共得到20个模块。本研究将与组织相关性高于0.65的模块定义为组织特异性模块, 最终鉴定到14个组织特异性模块。利用在线网站Agrigo对组织特异性模块中的基因进行GO (gene ontology)富集分析, 发现14个模块中均可以得到富集种类。开花作为玉米生育周期中的一个重要生理过程, 不仅代表着植物从营养生长到生殖生长的转变, 也关系到产量、株高和抗逆性等农艺性状。本研究发现8个组织特异性模块中的基因可以富集到与开花调控的代谢通路。此外, 有17个已经报道过的开花时间调控基因存在于共表达模块中, 并且主要分布在Blue模块和Darkmagenta模块, 因此本研究重点关注了这2个模块内部的基因调控网络。本研究通过计算不同组织中的基因表达丰度, 并联合权重基因共表达网络分析的方法, 鉴定到了具有生物学意义的共表达基因模块, 挖掘到了数个开花相关的模块, 有助于揭示玉米开花调控的遗传机制。
玉米; 权重基因共表达网络; 发育调控; 开花; 转录组
近年来, 随着高通量测序技术的飞速发展与成本不断的降低, 越来越多的研究人员采用多样本的转录组测序进行生物学问题的研究。用多样本的转录组数据可以系统地研究复杂条件下(例如多个组织、多个环境、多试验变量)的生物学问题, 然而传统的两个样本的相关性比较方法已经不能有效的处理海量的生物学数据。面对这个问题, 网络研究的方法脱颖而出, 当前生物学研究中广泛应用的网络研究方法有基因网络[1]、蛋白质调控网络[2]和代谢网络[3]等。借助于基因组学和转录组学的快速发展, 基因调控网络在理论和实践层面都取得了较快的发展。在基因调控网络中, 应用较广泛的是权重基因共表达网络。前人研究指出, 权重基因共表达网络内部的基因联通符合无尺度的网络拓扑结构, 即网络中少数基因作为网络中的核心节点与大部分基因具有联系, 该过程的实现依赖于对基因间的连接系数进行幂函数处理, 使网络逐渐趋向无尺度分布[4]。因此借助网络分析, 可以有效地利用海量的转录组数据, 将众多基因划分为相似的基因表达模块进行研究。权重基因共表达网络分析(weighted gene co-expression network analysis, WGCNA)根据基因表达水平的不同将基因聚类, 具有相似表达模式的基因被划分到同一个模块中。其目的在于研究共表达模块和目标性状之间的生物学相关性、鉴定组织特异性模块并研究模块内的核心基因。WGCNA作为一种系统生物学方法, 已经广泛应用于多样本的生物学问题研究当中。例如在动物中, 研究人员利用四个年龄段的恒河猴[5](L.), 分别选取8个组织进行转录组测序, 通过将不同的lncRNA和mRNA分为不同的模块, 阐释了lncRNA-lncRNA, lncRNA-mRNA基因间的共表达关系。WGCNA也广泛应用在植物研究中, 为了研究油菜(L.)在干旱处理条件下的mRNA的表达模式[6], 研究人员利用干旱和对照处理下的48个转录组数据进行了共表达网络构建, 进一步鉴定到与干旱处理相关的生物学模块, 并且挖掘了目标模块内的核心基因。在对草莓(L.)花的研究中[7], 研究者选取了6个组织, 12个发育时期的样品, 进行了转录组测序进而得到了共表达网络, 在特异性模块中鉴定到了大量的转录因子作为模块中的核心基因, 对促进花芽分化具有重要的作用, 此外该研究还表明WGCNA比K-MEAN聚类更具有特异性, 这是因为WGCNA构建的网络符合无尺度拓扑分布, 因此可以得到更多的具有生物学意义的模块。
玉米开花是一个多基因控制的数量性状, 代表了玉米从生殖生长到营养生长的转变。开花是玉米生育周期中的重要农艺性状, 关系到叶片数量、产量、营养价值以及抗逆性等性状。近年来, 研究人员利用分子生物学的方法, 已经在玉米中鉴定到了数十个开花调控基因。例如, 通过数量性状位点定位, 已经克隆了数个开花调控基因, 包括[8]、[9]和[10]。研究人员结合突变体分析和转基因技术鉴定到了在玉米中的同源基因[11], 研究结果表明是一个开花促进因子, 该基因在叶片中表达并通过韧皮部运送到玉米的顶端分生组织, 在顶端分生组织中与转录因子结合, 进而激活开花促进因子[12], 而的表达则标志着玉米开始从营养生长转变到生殖生长。此外, 有研究使用5000份重组玉米自交系进行巢式关联分析, 鉴定到数十个参与光周期调控的候选基因[13]。随着玉米参考基因组以及单倍型图谱的相继构建完成, 利用重测序、转录组和代谢组等生物信息学手段研究玉米的农艺性状已经成为一个有力的手段, 然而前人的研究着眼于单个调控因子(如顺式表达元件, 反式作用因子), 鲜有利用网络分析的方法解析玉米开花遗传机制的报道。
本研究利用玉米14个不同组织的RNA-seq数据, 计算得到全基因组水平上的基因表达量, 进而构建了具有高度拓扑性质的共表达网络, 得到了基因共表达模块, 并进一步筛选到了与组织高度关联的共表达模块。通过对组织特异性模块的基因富集分析发现了具有生物学功能的模块。此外, 结合富集分析的结果, 鉴定到了数个与开花调控相关的模块。本研究利用转录组数据得到了开花相关的共表达模块, 挖掘了模块内的开花相关的调控基因, 为玉米开花的遗传机制解析提供了新的研究思路和实验结果。
1 材料与方法
1.1 玉米转录组数据获取与分析
玉米不同发育阶段的转录组数据来源于NCBI(national center for biotechnology information)的SRA (Sequence Read Archive)数据库, 获取码为PRJNA171684[14]。转录组数据来源于叶片、顶端分生组织、花丝、花药、果穗等组织。首先利用SRAtoolkit软件的fast-dump参数将高通量测序数据转变为Fastq文件, 利用FastQC[15]软件对原始测序数据进行质量评估, 接着利用Trimmomatic软件去除接头、低质量和未知碱基数目过多的reads, 得到clean reads。参考Pertea流程[16], 利用Hisat2软件将clean reads比对到玉米B73参考基因组。玉米参考基因组序列和注释信息下载自ensemble数据库发布的玉米基因组信息(ftp://ftp.ensemblgenomes.org/ pub/plants/release-27/genbank/zea_mays/), 利用Stringtie软件对转录本组装, 之后采用R软件的Ballgown包计算基因在每一个组织中的转录本表达量。使用FPKM (fragments per kilobase of exon per million fragments mapped, 每千碱基外显子百万片段数)值来衡量基因的表达水平。
1.2 共表达网络构建
基因的表达谱矩阵来自所有样本的基因表达量。考虑到在各个组织中均低表达的基因不具有生物学意义, 在后续的计算中还会增加运算量, 因此为了保证共表达网络的特异性和快捷性, 基因在各个组织中的最大FPKM值小于5的将被过滤[17-18]。首先, 构建共表达网络需要计算每2个基因间的相关系数, 进而得到相似矩阵。使用公式表示如下: Smn=cor(xm, xn), S=[Smn], 式中Smn表示基因m和基因n之间的皮尔森相关系数, S表示相似矩阵。
R软件中的WGCNA包提供了一系列函数用来构建权重基因共表达网络[19-20]。为了使网络符合无尺度网络分布, 需要选择合适的权重值, 利用WGCNA包中的函数pickSoftThreshold计算权重值。根据图1的结果, 选择软阈值β=14用来构建共表达网络, 在该参数下每个基因的平均邻接系数是280, 中位数是165, 最大连接系数是1140。邻接矩阵的计算公式为Amn=[(1+Smn)/2]β, 为了清除背景噪音和伪关联带来的误差, 将邻接矩阵转为拓扑重叠矩阵(Topological Overlap Matrix, TOM), 同时利用函数dissTom=1–TOM对拓扑重叠矩阵取逆得到相异性矩阵。利用函数hclust对相异矩阵进行层次聚类。针对产生的聚类树, 采用动态切割 (Dynamic Tree Cut) 法切割基因聚类树, 相关参数为minModuleSize=30, 该过程可以把表达模式相近的基因合并在同一个分支, 每一个分支代表一个共表达模块(图2)。

图1 软阈值确定
图中的横轴均代表软阈值(β)。A: 纵坐标对应的是无尺度网络模型指数; B: 每一个软阈值对应的网络平均连接程度。
The abscissa represents the soft threshold (β). A: ordinate corresponds to the index of scale free network model; B: the average link degree of each soft threshold.
1.3 组织特异性模块鉴定
具有相似表达模式的基因被认为是具有类似的生物学功能。模块是一组表达模式高度相似的基因集合。为了深入研究与组织高度相关联的模块, 计算了模块特征向量(module eigengene, ME)和不同组织之间的相关系数, 相关系数越大, 该模块与组织间关联度越高, 而相关系数值越小, 模块与组织间关联度低。为了研究具有高度生物学特性的模块, 本研究定义相关系数的阈值为0.65, 即任何模块与组织的相关系数高于0.65将被定义为组织特异性模块[21]。
1.4 模块基因的富集分析
首先提取出组织特异性模块中的基因, 再将模块基因提交到Agrigo[22](GO Analysis Toolkit and Database for Agricultural Community)(http://bioinfo. cau.edu.cn/agriGO/index.php)在线分析工具, 采用奇异富集分析(singular enrichment analysis, SEA)法进行富集分析。使用玉米参考基因组B73作为参考数据库, 利用Fisher’s校验和Bonferronni多重校验来进行显著GO类型的筛选。
1.5 玉米开花调控通路的相关模块
开花作为一个复杂的农艺性状, 不仅与外界环境信号有关, 同时也涉及到内源性的信号调控[23]。根据已有的文献报道, 玉米开花受到日照、赤霉素以及DNA的甲基化修饰等因素的调控。利用开花调控相关的一些关键词例如开花(flower)、光(light)、赤霉素(gibberellin)、甲基化(methylation)、分生组织(meristem)等, 在组织特异性模块中查找参与调控开花的模块与基因。
1.6 基因互作网络的构建
根据拓扑重叠矩阵计算出模块中不同基因间的权重值, 权重值越高, 代表基因间的关联程度越高。筛选出模块间相关系数排名较高的基因, 同时挑选出模块中已经报道过的开花基因, 利用这些基因进行互作网络的构建, 网络中每一个节点代表一个基因, 连接线两端的基因具有相似的生物学功能。
2 结果与分析
2.1 基因共表达网络构建
通过数据分析共获得39,625个基因的表达谱数据, 过滤低表达量的基因, 获得22,426个高表达基因。计算得到了基因表达水平的样本聚类和性状关联, 图3的结果表明每一个组织的基因聚类树可以和组织很好地对应。通过对权重值筛选, 最终选择β=14来构建网络, 采用动态剪切树法合并表达相似的模块, 共获得20个共表达模块(图2), 不同的颜色代表不同的模块。模块中的基因数目分配根据其表达量进行一个相关度的聚类, 聚类度较高的基因被分配到一个模块中, 其中Black模块包含的基因数目最多, 是8492个基因, Floralwhite模块内的基因数量最少, 含87个基因, 平均每个模块包含的基因数目是1123, Grey模块是一组未分配到其他模块的基因集合(图2)。

图2 基因聚类树和模块切割
A: 基于拓扑重叠构建的基因聚类树。B: 动态混合切割法得到的基因模块, 颜色代表模块。C: 合并相似表达模式的基因模块。
A: clustering of genes based on the topological overlap. B: the gene modules obtained from the dynamic tree cut. C: the merged modules with similar expression pattern.
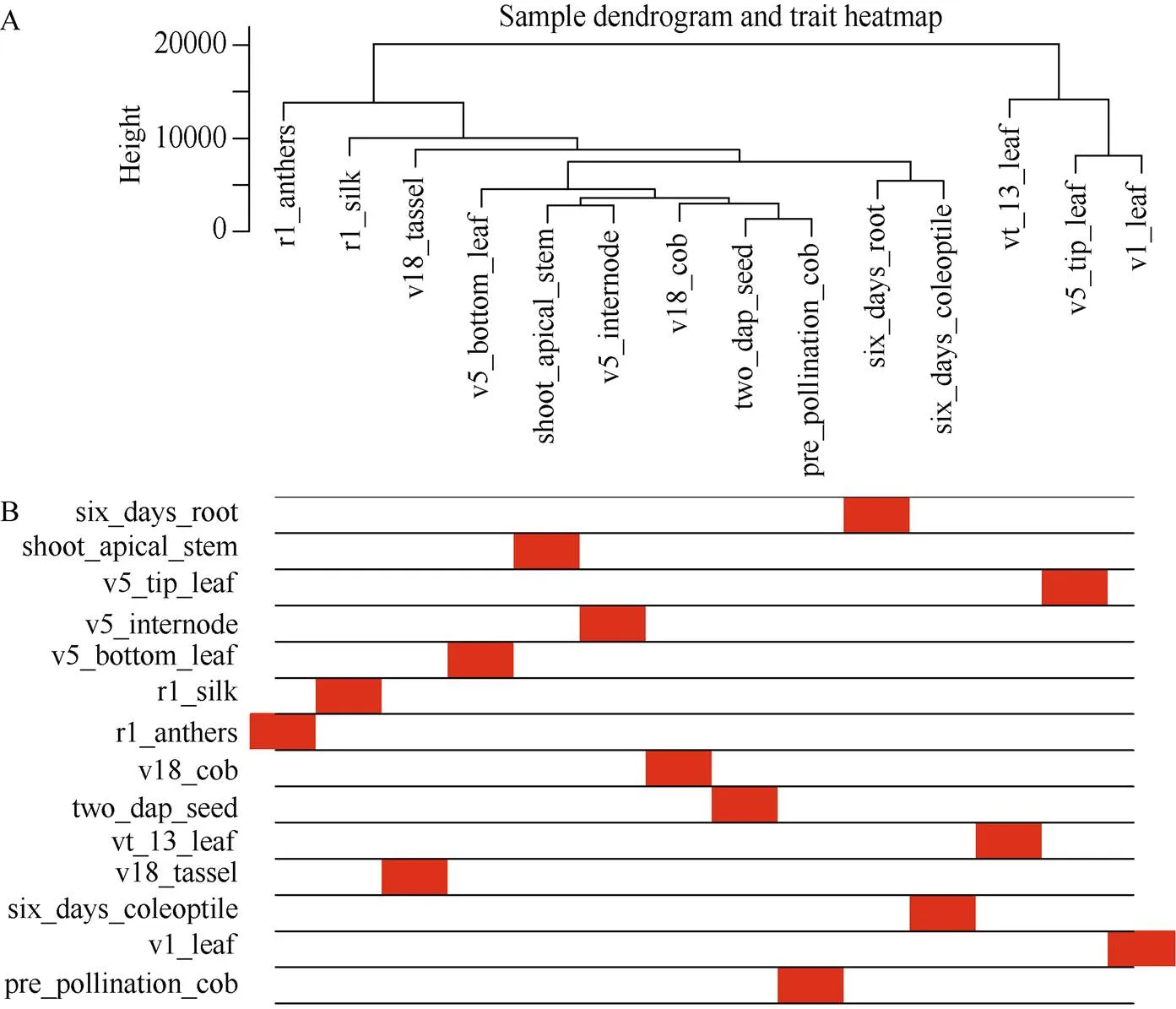
图3 样本层次聚类树及对应的组织信息
A: 基于欧氏距离得到的基因聚类树。横轴表示不同的玉米组织, 纵轴代表基因间的聚类高度。B: 性状与基因聚类树关联热图。白色的代表低关联度, 红色的代表高关联度。
A: the gene cluster tree based on the Euclidean distance. The abscissa represents the different tissues. The ordinate represents the cluster height of genes. B: the association trait heatmap, the white means a low correlation, the red means a high correlation.
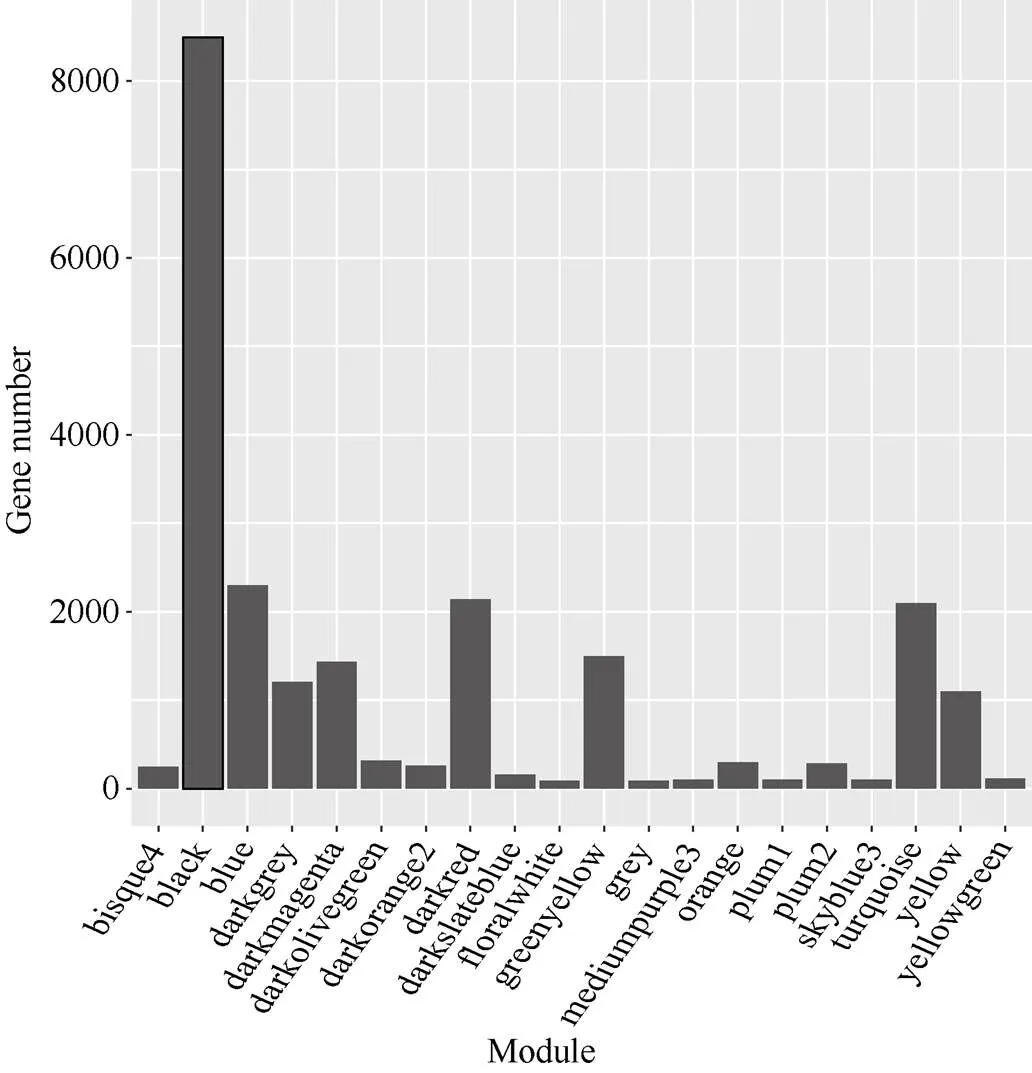
图4 共表达模块中基因数目分布
横轴代表模块, 纵轴代表模块中的基因数目。
The abscissa represents the modules and the vertical ordinate represents the gene number of each module.
2.2 组织特异性模块鉴定
在20个模块中有14个与组织存在高度特异性(图5)。大多数组织都有与其高度相关联的模块, 例如Blue模块与13叶期叶片存在高度相关性(=0.96,=6E–08), Darkmagenta与v18雄穗连接度较高(=0.98,=2E–09)。14个组织特异性模块中没有任何一个模块和v18未成熟果穗与授粉前果穗两个组织存在高度相关性。我们推测这两个时期组织中的基因主要进行常规的生命活动, 因此没有组织特异性的模块与之对应。
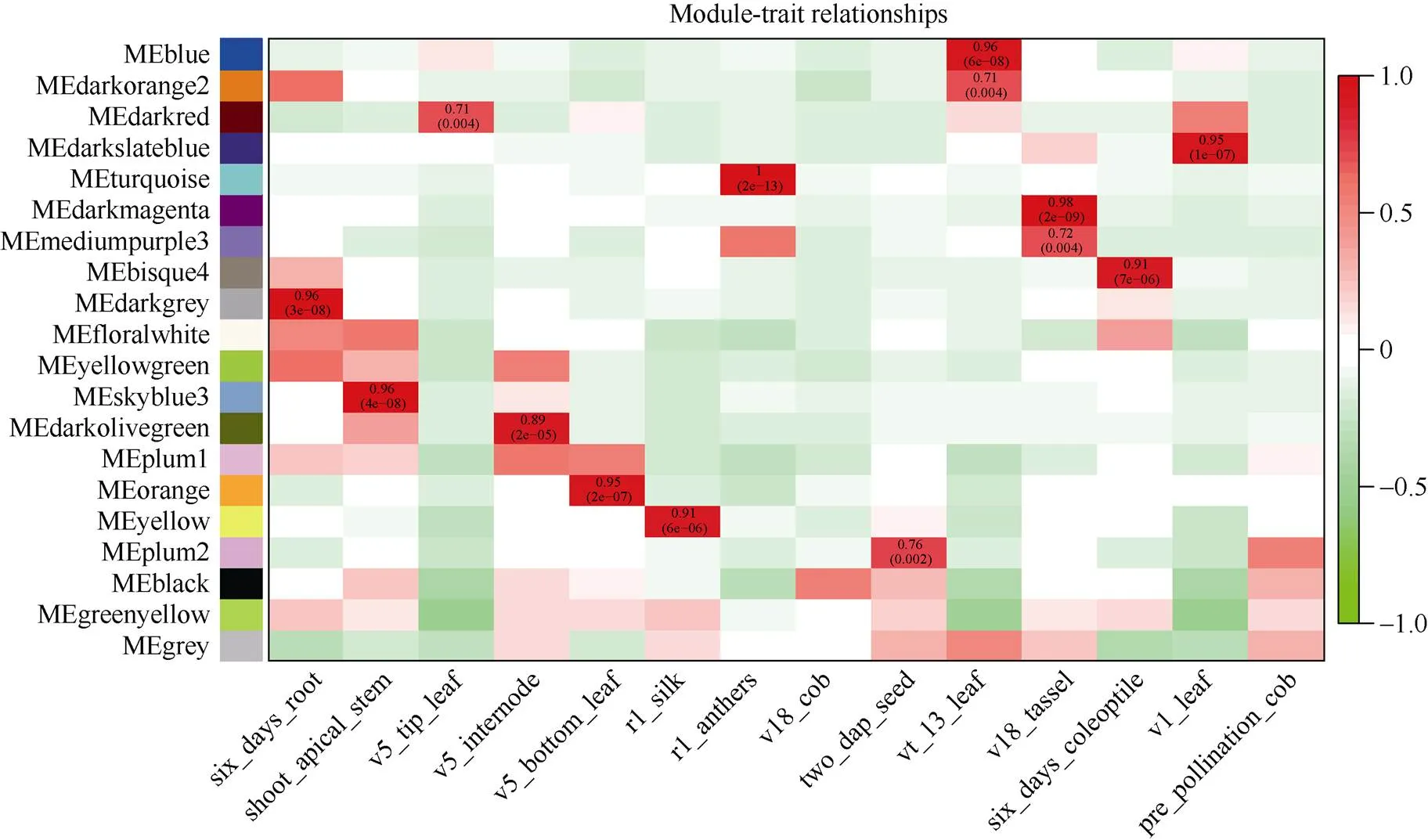
图5 模块与性状关联热图
横轴表示不同的性状, 纵轴表示每一个模块的特征向量。红色的格子代表性状与模块具有正相关性, 绿色的格子代表性状与模块具有负相关性。
Each column represents the co-expression module. Red color of each box represents the positive correlation between module and trait. Green color of each box represents the negative relationships between module and trait.
2.3 组织特异性模块的功能富集分析
具有相似表达模式的基因可能具有相同的生物学功能, 通过共表达网络构建的模块在很大程度上与特定的生物组织相联系, 而特定的生物组织又与特定生命活动存在高度协同性[4]。为了进一步解析组织特异性模块的生物学功能, 本研究利用Agrigo工具对模块基因进行富集分析, 表明这14个模块都可以富集得到显著的GO通路, 这些GO通路涉及到生物学过程(biological process, BP), 分子功能 (molecular function, MF)以及细胞组分(cellular component, CC)。与根发育高度关联Darkgrey模块可以富集得到水转运(GO:0006833), 流体运送(GO:0042044)等调控通路, 表明WGCNA可以构建到具有生物学意义的共表达模块。通过对开花发育与调控的关键字进行检索发现, Blue、Darkmagenta、Darkred、Darkslateblue、Orange、Plum2、Yellow和Bisuqe4模块都富集到了与开花相关的调控通路(图6), 这些通路主要是响应光照刺激的调控通路(GO:0009416)、顶端分生组织形态保持(GO:0010073)和花器官发育(GO:0048437)等。Blue模块与13叶期叶片存在最高的相关性(=0.96,=6E–08), Darkmagenta模块和v18雄穗具有最高的相关性(=0.98,=2E–09), 这两个模块对应的组织是叶片和雄穗, 是和开花密切相关的组织, 此外, Blue模块包含了7个已经报道的开花基因, Darkmagenta模块也包含6个已经报道的开花基因, 虽然富集得到了开花调控通路, 但是开花基因数目较少或者没有已知开花基因存在于这些模块, 因此本研究重点关注了Blue模块(图7)和Darkmagenta (图8)模块, 筛选出模块中高连通性的基因, 利用Cytoscape[24]软件对网络进行了可视化处理, 处于网络中的基因是该模块中的高连接度的基因。
2.4 开花基因互作网络构建
考虑到开花时间调控基因主要分布在Blue和Darkmagenta模块, 因此利用这2个模块中已经报道的开花基因构建基因互作网络。Blue模块中共发现7个报道过的开花基因, 其中、[25]和[26]3个基因的连通性在模块排名为前1%, 可以作为模块中的核心基因,和作为开花促进因子, 是开花调控通路中的重要基因。此外[27]、[28-29]、[30]和[31]基因在模块中联通性排名靠后, 但是这些基因被证明是玉米开花调控通路中关键基因, 推测WGCNA构建的模块中, 基因的连通性基于基因间表达量, 而基因间表达量受多种因素影响, 导致关键调控基因在模块中的相关性较低。因此, 本研究以、、、、、、等基因作为枢纽基因, 筛选与开花基因高连通性的基因, 将其定义为候选开花基因, 利用开花基因和候选花期基因构建局部调控网络(图9), 在该网络中, 每一个节点代表一个基因, 节点间通过连接线相联系, 处于连接线两端的基因被认为具有相同生物功能。与v18雄穗高度关联的Darkmagenta模块包含6个开花调控基因, 即[32]、[33]、[32]、[34]、[34]和[35], 在该模块中有2个基因的连通性排名前10%, 分别是和, 参照Blue模块的研究思路, 利用Darkmagenta已经报道过的开花基因构建Darkmagenta模块内的局部调控网络(图10)。此外, 检索了候选开花基因在拟南芥中的同源基因, 并借助TAIR (https://www.arabidopsis.org/)网站注释了拟南芥同源基因的功能(表1)。
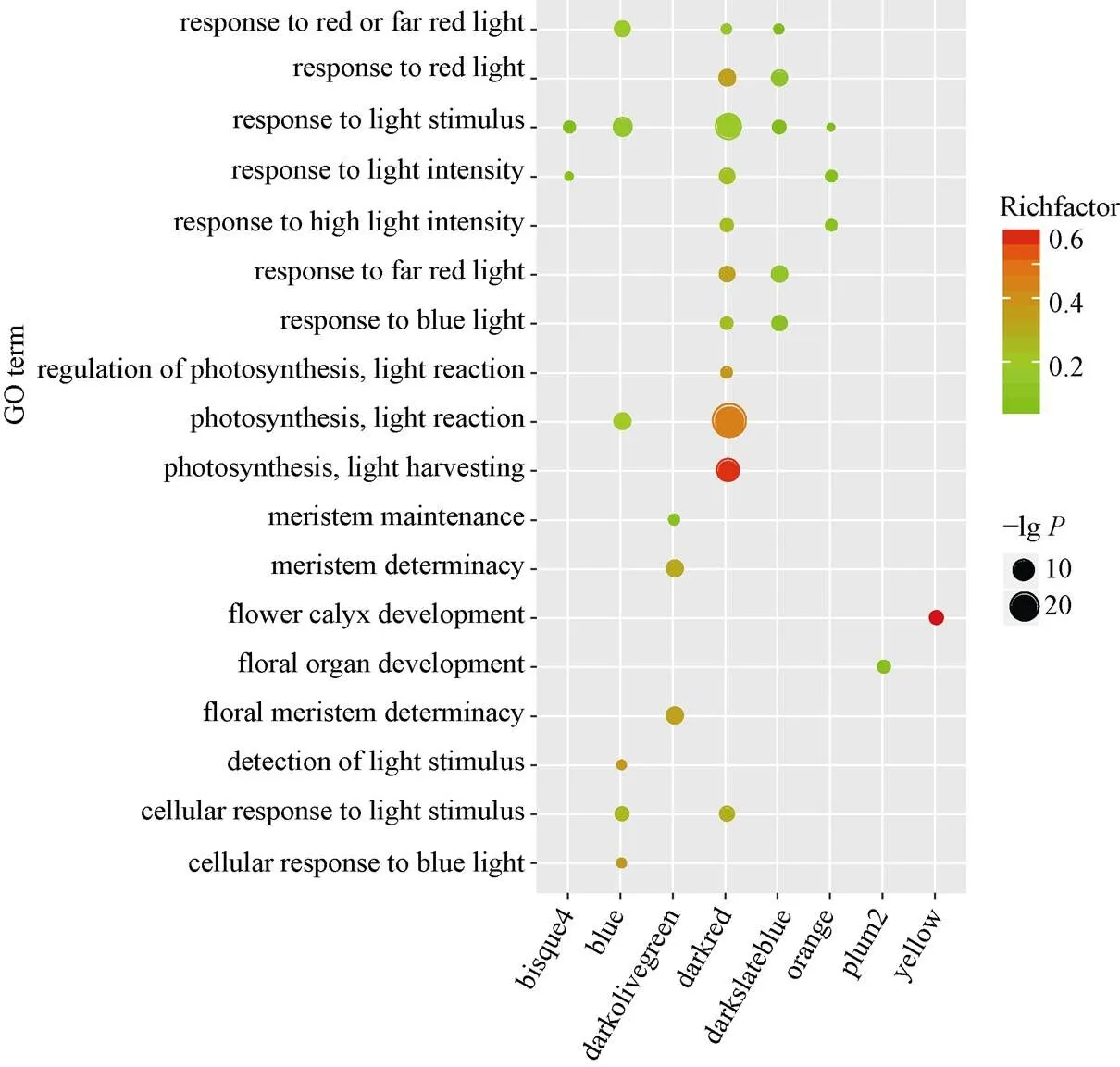
图6 开花富集通路
每一行代表开花富集通路, 每一列代表组织特异性模块。点的大小代表多重校验的值大小, 点的颜色表示输入基因与背景基因的比值。
Each row corresponds to a GO enrichment pathway, column to a tissue specific module. The point size is calculated by the-value of multiple check. The Richfactor represent the ratio of input genes to the background genes.

图7 Blue模块内的基因共表达网络

图8 Darkmagenta模块内基因共表达网络
3 讨论
传统的生物学研究侧重于从分子水平阐释单个功能元件(如DNA、mRNA和蛋白质)对生命活动的影响, 虽然该方法对于揭示具体性状的遗传机理具有非常重要的意义, 但只能局部地解释某一生命活动发生的原因。随着测序技术的快速发展, 传统的生物学研究不能充分有效地挖掘海量数据中蕴含的生物学意义。网络作为系统生物学的一种研究手段, 借助基因组、转录组和代谢组的数据, 广泛地应用于生命科学的探索中。相比于其他调控网络, WGCNA可以特异地筛选出与性状相关的基因, 并进行模块化分类, 得到具有高度生物学意义的共表达模块, 已经被证明是一种高效的数据挖掘手段[36]。
本研究通过对14个组织特异性模块进行富集分析, 发现这14个模块均可以得到具有生物学意义的调控通路(附表1)。例如在与根高度关联的Darkgrey模块, 富集分析的GO term包含水转运(GO:0006833)、流体运输(GO:0042044)等通路, 这些通路和根部所具有的生物功能高度吻合, 该结果证明了共表达网络构建模块的特异性。本研究直接利用转录组数据对应的组织, 例如根、叶片和花药等作为研究的性状, 该方法在前人的研究中已有过相关报道, 如在黄瓜(L.) 10个组织的共表达网络构建中, 研究人员成功鉴定到数个与黄瓜苦味合成代谢相关的模块[18]。
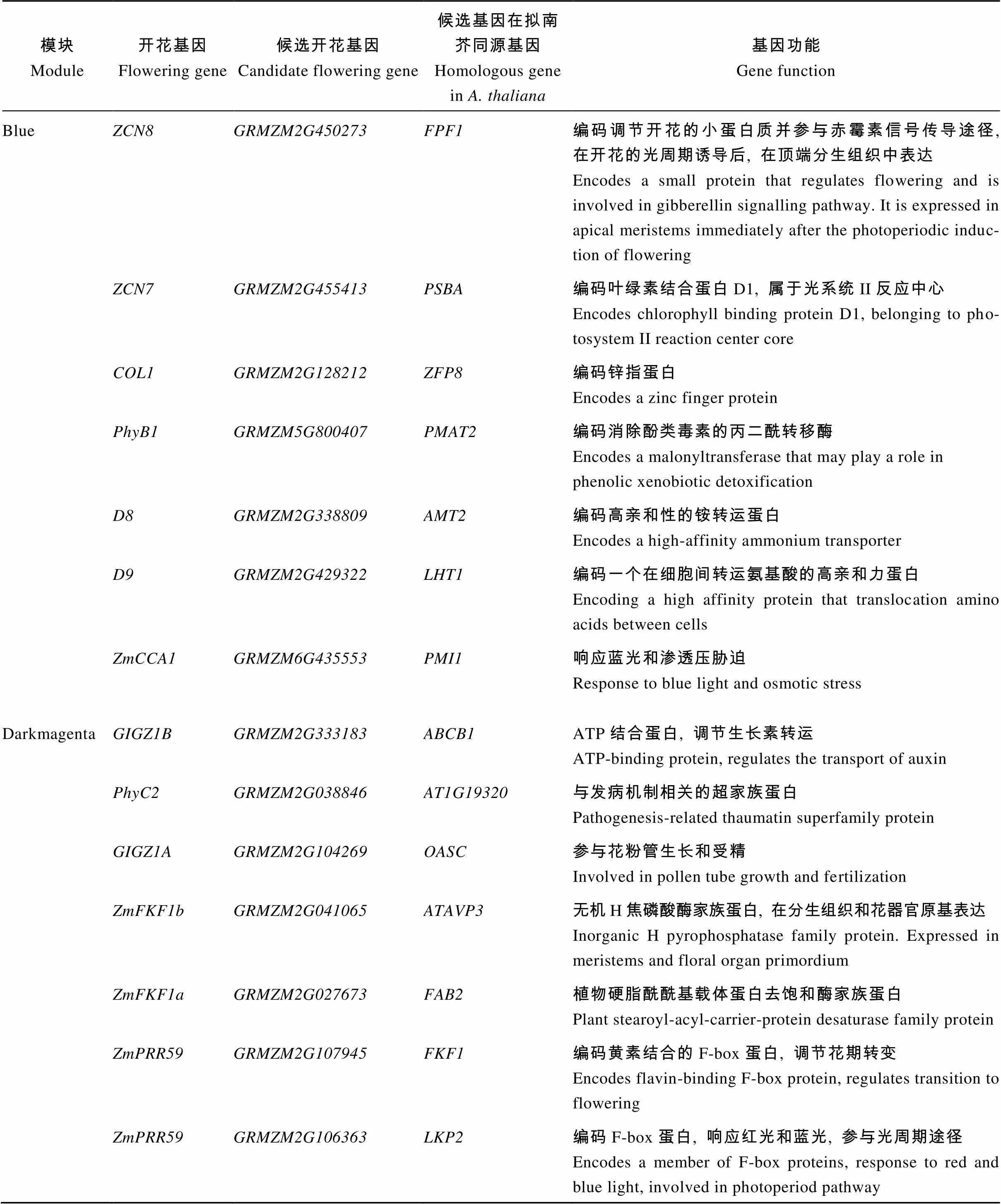
表1 Darkmagenta模块和Blue模块中候选开花基因的功能注释。
本研究重点关注了开花这个重要的农艺性状。开花作为玉米生命活动的中心环节, 对其遗传机制的解析虽然取得了一定的进展, 例如通过数量性状位点定位和关联分析等方法鉴定了许多重要的开花基因, 但是这些研究手段都有一定的局限性, 例如数量性状定位不能反映参考群体较为广泛的遗传异质性[37], 此外, 许多复杂的农艺性状是一个与群体结构存在高度相关的性状, 例如开花, 而且利用关联分析鉴定控制目标性状相关的调控基因容易受到群体结构的影响[38]。权重基因共表达网络则通过共表达模块化处理, 将成千上万的基因分配到模块中, 通过研究模块蕴含的生物学意义进一步挖掘相应基因的功能, 因此较好地解决了对复杂性状解析能力不足的问题, 被广泛地应用于基因组学的研究之中。

图9 Blue模块开花相关的基因网络
红色节点是参与开花调控通路的基因。
Each node corresponding to a gene, the red node is the hub gene of the network and is the flowering time gene which had reported.
在本研究中的富集分析结果中, 8个组织特异性模块包含开花的调控通路(图6), 例如Blue模块中的响应光刺激调控通路(GO:0009416), Darkolivegreen模块中的顶端分生组织形态维持(GO:0010022)和Plum2模块的花器官发育(GO:0048437)。此外, 结合已经报道过的开花基因, 进一步发现Blue模块和Darkmagenta模块含有较多开花相关的调控基因, 而且这些开花基因的连通性在模块中很高, 处于枢纽地位, 例如Blue模块中的和基因是开花激活因子, Darkmagenta模块中的和基因, 是时钟通路的重要参与基因。通过对模块中基因的局部网络化, 挖掘到了与开花基因连接程度较高的基因, 这些基因可以作为开花候选基因被进一步研究。例如在Blue模块中, 与8高连通性的基因, 其在拟南芥中的同源基因是,基因编码一个调节开花的小蛋白质, 并且参与赤霉素调控通路, 在光照诱导后该基因在顶端分生组织表达,的高连接度基因在拟南芥中的同源基因是, 通过功能注释发现这个基因编码叶绿素结合蛋白D1, 属于光系统II反应中心, 参与光周期调控通路。在Darkmagenta模块中,的高连接度基因是, 其在拟南芥中的同源基因是, 该基因编码一个F-box蛋白, 响应光照刺激, 参与光周期途径;是的高连接度基因, 该基因在拟南芥中的同源基因是, 主要参与花粉管生长和受精。以上结果表明, 在WGCNA中, 与目标基因存在高连通性的基因具有与其相似的生物学意义, 这可以为挖掘目标基因提供新的研究思路。在本研究中, 重点关注了Blue和Darkmagenta两个开花相关的调控模块, 其余6个开花相关的基因模块虽然没有被详细的论述, 但是其包含开花富集通路, 可进一步挖掘其蕴含的生物学意义。
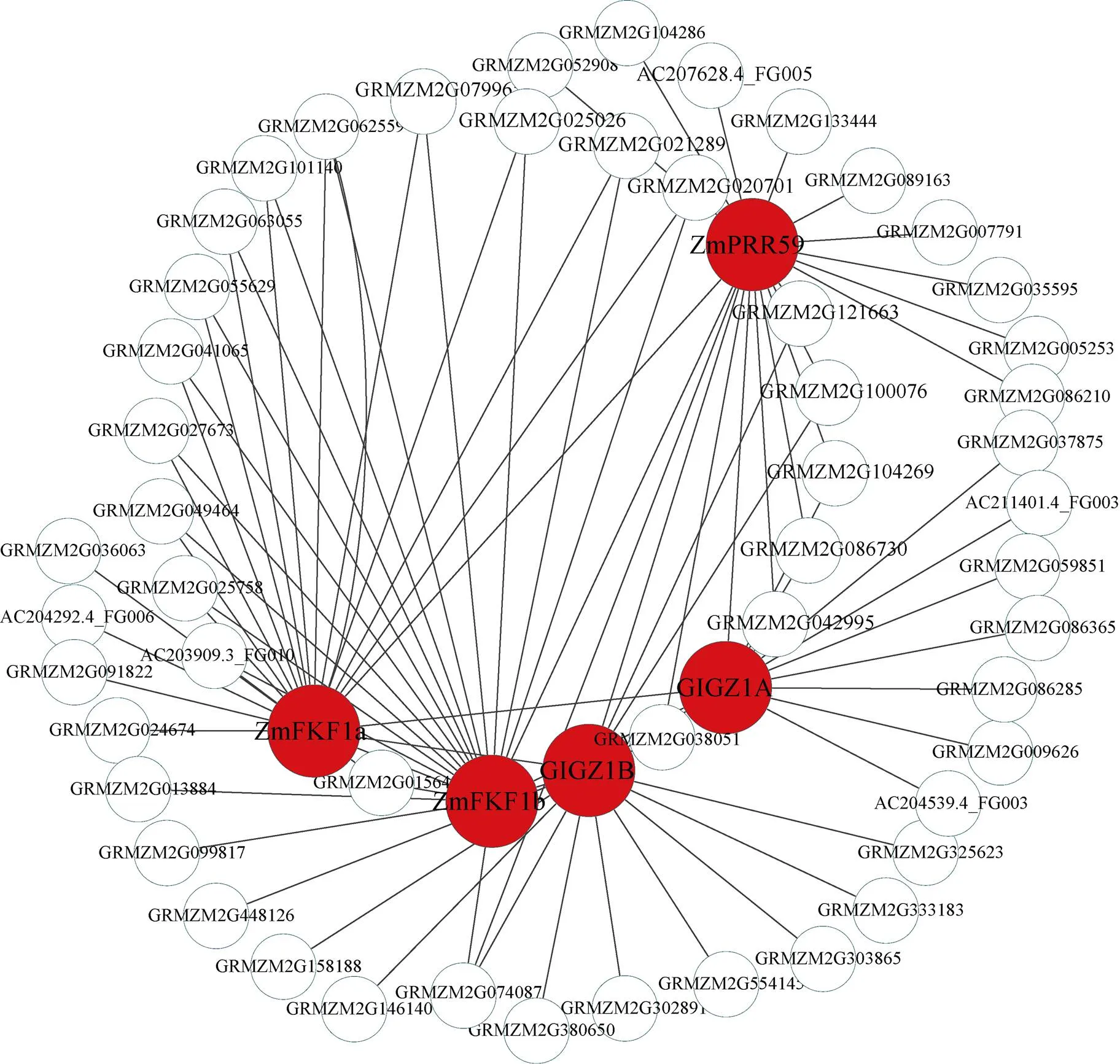
图10 Darkmagenta模块的开花相关的基因网络
红色节点基因参与开花调控通路。
Each node corresponding to a gene, the red node is the hub gene of the network and is the flowering time gene which had reported.
本研究鉴定到的组织特异性模块对应的组织都是玉米开花调控和发育的重要参与器官, 因此, 利用共表达网络解析具体的农艺性状, 分析模块的生物功能, 挖掘模块中的目标基因, 将为解析复杂的农艺性状提供重要的参考依据。
4 结论
构建了与性状相关联的权重基因共表达网络, 得到了14个组织特异性模块。揭示了组织特异性模块蕴含的生物学意义, 鉴定了开花相关的调控基因, 并用其构建了局部的具有生物学意义的网络。本研究结果可为后续解析玉米发育的遗传机理提供重要的实验数据和理论基础。
[1] Stuart J M, Segal E, Koller D, Kim S K. A gene-coexpression network for global discovery of conserved genetic modules., 2003, 302: 249–255.
[2] Jeong H, Mason S P, Barabási A L, Oltvai Z N. Lethality and centrality in protein networks., 2001, 411: 41.
[3] Gille C, Hoffmann S, Holzhütter H G. METANNOGEN: compiling features of biochemical reactions needed for the reconstruction of metabolic networks., 2007, 1: 5.
[4] Barabási A L, Oltvai Z N. Network biology: understanding the cell's functional organization., 2004, 5: 101–113.
[5] Liu S, Wang Z, Chen D, Zhang B, Tian R R, Wu J, Zhang Y, Xu K Y, Yang L M, Cheng C, Ma J, Lv L B, Zheng Y T, Hu X T, Yi Z, Wang X T, Li J L. Annotation and cluster analysis of spatiotemporal- and sex-related lncRNA expression in rhesus macaque brain., 2017, 27: 1608–1620.
[6] Greenham K, Guadagno C R, Gehan M A, Mockler T C, Weinig C, Ewers B E, McClung C R. Temporal network analysis identifies early physiological and transcriptomic indicators of mild drought in., 2017, 6: e29655.
[7] Hollender C A, Kang C, Darwish O, Geretz A, Matthews B F, Slovin J, Alkharouf N, Liu Z. Floral transcriptomes in woodland strawberry uncover developing receptacle and anther gene networks., 2014,165: 1062–1075.
[8] Vlăduţu C, McLaughlin J, Phillips R L. Fine mapping and characterization of linked quantitative trait loci involved in the transition of the maize apical meristem from vegetative to generative structures., 1999, 153: 993–1007.
[9] Wong A Y, Colasanti J. Maize floral regulator protein INDETERMINATE1 is localized to developing leaves and is not altered by light or the sink/source transition., 2007, 58: 403–414.
[10] Muszynski M G, Dam T, Li B, Shirbroun D M, Hou Z, Bruggemann E, Archibald R, Ananiev E V, Danilevskaya O N. Delayed flowering1 encodes a basic leucine zipper protein that mediates floral inductive signals at the shoot apex in maize., 2006, 142: 1523–1536.
[11] Meng X, Muszynski M G, Danilevskaya O N. The-likegene functions as a floral activator and is involved in photoperiod sensitivity in maize., 2011, 23: 942–960.
[12] Danilevskaya O N.encodes a basic leucine zipper protein that mediates floral inductive signals at the shoot apex in maize., 2006, 142: 1523–1536.
[13] Coles N D, McMullen M D, Balint-Kurti P J, Pratt R C, Holland J B. Genetic control of photoperiod sensitivity in maize revealed by joint multiple population analysis., 2010, 184: 799–812.
[14] Sekhon R S, Lin H, Childs K L, Hansey C N, Buell C R, de Leon N, Kaeppler S M. Genome-wide atlas of transcription during maize development., 2011, 66: 553–563.
[15] Kroll K W, Mokaram N E, Pelletier A R, Frankhouser D E, Westphal M S, Stump P A, Stump C L, Bundschuh R, Blachly J S, Yan P. Quality Control for RNA-Seq (QuaCRS): an integrated quality control pipeline., 2014, 13: 7–14.
[16] Pertea M, Kim D, Pertea G M, Leek J T, Salzberg S L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown., 2016, 11: 1650–1667.
[17] 魏凯, 张婷婷, 马磊. 猪基因共表达网络模块的构建及功能分析. 畜牧兽医学报, 2017, 48: 2205–2215.Wei K, Zhang T T, Ma L. Construction and functional analysis of gene co-expression network modules., 2017, 48: 2205–2215 (in Chinese with English abstract).
[18] 林行众, 张忠华, 杨清, 黄三文. 黄瓜共表达基因模块的识别及其特点分析. 农业生物技术学报, 2017, 23: 1121–1130. Lin X Z, Zhang Z H, Yang Q, Huang S W. Identification and characterization analysis of co-expression gene modules in cucumber (L.)., 2017, 23: 1121–1130 (in Chinese with English abstract).
[19] Zhang B, Kirov S, Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts., 2005, 33: W741–W748.
[20] Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis., 2008, 9: 559.
[21] Downs G S, Bi Y M, Colasanti J, Wu W, Chen X, Zhu T, Rothstein S J, Lukens L N. A developmental transcriptional network for maize defines coexpression modules., 2013, 161: 1830–1843.
[22] Du Z, Zhou X, Ling Y, Zhang Z H, Su Z. agriGO: a GO analysis toolkit for the agricultural community., 2010, 38: 64–70
[23] Dong Z S, Danilevskaya O, Abadie T, Messina C, Coles N, Cooper M. A gene regulatory network model for floral transition of the shoot apex in maize and its dynamic modeling., 2012, 7: e43450.
[24] Shannon P, Markiel A, Ozier O, Baliga N S, Wang J T, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks., 2003, 13: 2498–2504.
[25] Mascheretti I, Turner K, Brivio R S, Hand A, Colasanti J, Rossi V. Florigen-Encoding genes of day-neutral and photoperiod- sensitive maize are regulated by different chromatin modifications at the floral transition., 2015, 168: 1351–1363.
[26] Khan S, Rowe S C, Harmon F G. Coordination of the maize transcriptome by a conserved circadian clock., 2010, 10: 126.
[27] Sheehan M J, Kennedy L M, Costich D E, Brutnell T P. Subfunctionalization ofandin the control of seedling and mature plant traits in maize., 2007, 49: 338–353.
[28] Thornsberry J M, Goodman M M, Doebley J, Kresovich S, Nielsen D, Buckler E S. Dwarf8 polymorphisms associate with variation in flowering time., 2001, 28: 286.
[29] Larsson S J, Lipka A E, Buckler E S. Lessons fromon the strengths and weaknesses of structured association mapping., 2013, 9: e1003246.
[30] Lawit S J, Wych H M, Xu D, Kundu S, Tomes D T. Maize DELLA proteins dwarf plant8 and dwarf plant9 as modulators of plant development., 2010, 51: 1854–1868.
[31] Wang X, Wu L, Zhang S, Wu L, Ku L, Wei X, Xie L, Chen Y. Robust expression and association ofwith circadian rhythms in maize., 2011, 30: 1261–1272.
[32] Miller T A, Muslin E H, Dorweiler J E. A maize CONSTANS-like gene,, exhibits distinct diurnal expression patterns in varied photoperiods., 2008, 227: 1377–1388.
[33] Sheehan M J, Farmer P R, Brutnell T P. Structure and expression of maize phytochrome family homeologs., 2004, 167: 1395–1405
[34] Hayes K R, Beatty M, Meng X, Simmons C R, Habben J E, Danilevskaya O N. Maize global transcriptomics reveals pervasive leaf diurnal rhythms but rhythms in developing ears are largely limited to the core oscillator., 2010, 5: e12887.
[35] Chardon F, Virlon B, Moreau L, Falque M, Joets J, Decousset L, Murigneux A, Charcosset A. Genetic architecture of flowering time in maize as inferred from quantitative trait loci meta- analysis and synteny conservation with the rice genome., 2004, 168: 2169–2185.
[36] Zhao W, Langfelder P, Fuller T, Dong J, Li A, Hovarth S. Weighted gene coexpression network analysis: state of the art., 2010, 20: 281–300.
[37] Holland J B. Genetic architecture of complex traits in plants., 2007, 10: 156–161.
[38] Camus-Kulandaivelu L, Veyrieras J B, Madur D, Combes V, Fourmann M, Barraud S, Dubreuil P, Gouesnard B, Manicacci D, Charcosset A. Maize adaptation to temperate climate: relationship between population structure and polymorphism in thegene., 2006, 172: 2449–2463.
附表1 组织特异性模块的GO富集分析结果(部分)
Supplementary table 1 GO enrichment analysis results of tissue specific module (part)

模块ModuleGO条目GO term本体Ontology描述DescriptionP值P-value Darkorange2GO: 0009889BP生物合成过程调节Regulation of biosynthetic process6.90E–07 Darkorange2GO: 0006355BPDNA依赖的转录调节Regulation of transcription, DNA-dependent4.40E–07 Darkorange2GO: 0015267MF通道活性Channel activity0.00068 Darkorange2GO: 0022838MF底物特异性通道活性Substrate-specific channel activity0.00068 BlueGO: 0009628BP非生物刺激的反应Response to abiotic stimulus3.60E–10 BlueGO: 0009416BP光刺激响应Response to light stimulus1.60E–08 BlueGO: 0003700MF转录因子活性Transcription factor activity0.00017 BlueGO: 0009535CC叶绿体类囊体膜Chloroplast thylakoid membrane0.00022 DarkredGO: 0051186BP辅因子代谢过程Cofactor metabolic process1.20E–19 DarkredGO: 0004252MF丝氨酸型肽链内切酶活性Serine-type endopeptidase activity0.00015 DarkredGO: 0009543CC叶绿体类囊体腔 Chloroplast thylakoid lumen4.40E–13 DarkslateblueGO: 0016051BP碳水化合物生物合成过程Carbohydrate biosynthetic process5.60E–05 DarkslateblueGO: 0016830MF碳-碳裂解酶活性Carbon-carbon lyase activity3.40E–06 DarkslateblueGO: 0042651CC类囊体膜Thylakoid membrane0.00013 TurquoiseGO: 0060560BP形态发生发育Developmental growth involved in morphogenesis1.70E–11 TurquoiseGO: 0015299MF溶质: 氢反向转运蛋白活性Solute: hydrogen antiporter activity4.60E–05 TurquoiseGO: 0031224CC内膜Intrinsic to membrane8.50E–06 DarkmagentaGO: 0010927BP涉及形态发生的内膜组装Cellular component assembly involved in morphogenesis4.10E–09 DarkmagentaGO: 0004553MF水解酶活性, 水解O-糖基化合物Hydrolase activity, hydrolyzing O-glycosyl compounds7.60E–06 DarkmagentaGO: 0030312CC高尔基体Golgi apparatus0.0004 Bisque4GO: 0042180BP细胞酮代谢过程Cellular ketone metabolic process3.00E–08 Bisque4GO: 0044281BP小分子代谢过程Small molecule metabolic process9.90E–06 Bisque4GO: 0042221BP相应化学刺激Response to chemical stimulus0.00032 DarkgreyGO: 0010033BP对有机物质的反应Response to organic substance1.60E–07 DarkgreyGO: 0004553MF水解酶活性, 水解O-糖基化合物Hydrolase activity, hydrolyzing O-glycosyl compounds4.40E–10 DarkgreyGO: 0005740CC线粒体包膜Mitochondrial envelope7.40E–05 FloralwhiteGO: 0006412BP蛋白质翻译Translation9.10E–07 FloralwhiteGO: 0005198MF结构分子活性Structural molecule activity5.80E–08 FloralwhiteGO: 0043227CC膜有界细胞器Membrane-bounded organelle0.0085 DarkolivegreenGO: 0010022BP分生组织决定Meristem determinacy6.10E–07
(附续表1)

模块ModuleGO条目GO term本体Ontology描述DescriptionP值P-value DarkolivegreenGO: 0003700MF转录因子活性Transcription factor activity5.50E–05 OrangeGO: 0042542BP过氧化氢反应Response to hydrogen peroxide2.90E–05 OrangeGO: 0009526CC质体Plastid9.60E–09 YellowGO: 0009605BP响应外界刺激Response to external stimulus8.40E–10 YellowGO: 0022892MF底物特异性转运体活性Substrate-specific transporter activity0.00013 Plum2GO: 0048437BP花器官发育Floral organ development9.20E–05 Plum2GO: 0005576CC胞外区Extracellular region9.70E–05 GreenyellowGO: 0034220BP离子跨膜转运Ion transmembrane transport5.00E–13 GreenyellowGO: 0032561MF鸟苷酸核糖核酸结合Guanyl ribonucleotide binding2.40E–10 GreenyellowGO: 0043231CC内细胞器膜Organelle inner membrane1.20E–17
Identification of maize flowering gene co-expression modules by WGCNA
YANG Yu-Xin1, SANG Zhi-Qin1,2, XU Cheng1, DAI Wen-Shuang1, and ZOU Cheng1,*
1Institute of Crop Sciences, Chinese Academy of Agricultural Sciences, Beijing 100081, China;2Xinjiang Academy of Agricultural and Reclamation Science, Shihezi 832000, Xinjiang, China
Weighted gene co-expression network analysis (WGCNA) is one of the research methods in systematic biology. It can effectively analyze the complex samples, and has been extensively used in the analysis of complicated traits for many samples. Weighted gene co-expression network has the characteristics of scale-free distribution and could construct the scale free network. The genes with similar expression level can be clustered and assigned to a module, then the relationships between co-expression modules and specific tissues can be furtherly analyzed. Our research utilized the transcriptome data of 14 different tissues of maize (L.) inbred line B73, and calculated the gene expression level of the whole genome. Through filtering out the genes with low expression level we finally got 22,426 genes with high expression level to construct the gene expression matrix. We utilized the different tissues as the trait to construct the trait matrix. The weighted gene co-expression network analysis packages of R software was used to perform the co-expression network analysis, and 20 co-expression modules were identified. We finally obtained 14 tissue specific modules which were highly correlated with traits (> 0.65). The enrichment analysis tool Agrigo was taken to perform the GO enrichment of the tissue specific module genes, all the 14 tissues could be enriched in GO terms. Flowering is one of the important agronomic traits in the life cycle of maize controlled by external environment signals and genetic factors. Maize flowering not only represents the transition from the vegetative growth to reproductive growth, also relates to grain yield, plant height and resistance. In our research, we detected eight tissue specific modules, which could be obtained within flowering time related pathways. In addition, 17 flowering genes which have been reported in the literatures were assigned to the co-expression modules, and mainly assigned to the Blue and Darkmagenta modules. Therefore, we focused on the network of Blue and Darkmagenta modules. Our research calculated the gene expression abundances, and detected several flowering time related modules, which will contribute to revealing the genetic mechanism of maize flowering time regulation.
maize; weighted gene co-expression network; development regulation; flowering; transcriptome
2018-07-17;
2018-10-08;
2018-11-08.
10.3724/SP.J.1006.2019.83053
邹枨,E-mail: zoucheng@caas.cn
E-mail: yyx0719@126.com
本研究由国家重点研发计划项目(2016YFD0100303)和国家自然科学基金项目(31371638)资助。
This study was supported by the National Key Research and Development Program of China (2016YFD0100303), the National Natural Science Foundation of China (31371638).
URL:http://kns.cnki.net/kcms/detail/11.1809.S.20181105.0933.004.htm
猜你喜欢
现代仪器与医疗(2022年4期)2022-10-08
中国农业科学(2022年11期)2022-06-27
健康体检与管理(2022年4期)2022-05-13
安徽农业科学(2022年1期)2022-02-14
中国饲料(2021年17期)2021-11-02
现代临床医学(2021年5期)2021-11-02
现代畜牧科技(2021年4期)2021-07-21
江苏农业学报(2021年2期)2021-06-30
中国生殖健康(2020年2期)2021-01-18
福建基础教育研究(2019年1期)2019-09-10
