
基于重测序的陆地棉InDel标记开发与评价
2019-01-17 01:28温天旺林忠旭
作物学报 2019年2期
吴 迷 汪 念 沈 超 黄 聪 温天旺 林忠旭
基于重测序的陆地棉InDel标记开发与评价
吴 迷 汪 念 沈 超 黄 聪 温天旺 林忠旭*
华中农业大学植物科学技术学院 / 作物遗传改良国家重点实验室, 湖北武汉 430070
碱基插入/缺失(InDel)是基因组中丰富的遗传变异形式。InDel以其密度高、易于基因型分型等优点成为分子标记开发的理想来源。本研究利用262份陆地棉品系重测序数据鉴定的InDel位点, 在全基因组范围内设计了3206个InDel标记并挑选均匀分布的320个标记进行验证。320个标记筛选出87个多态性标记, 多态性率为26.88%。利用多态性标记对不同地理来源的262份陆地棉种质资源进行基因分型, 共检测到160个等位位点; 多态性信息含量(PIC)为0.0836~0.3750, 平均值为0.3073; 基因多样性指数变异范围为0.0874~0.5000, 平均值为0.3876, 表明我国陆地棉遗传基础相对狭窄。群体结构分析将262份陆地棉品系大致划分为2个亚群, 聚类分析和主成分分析的结果与之基本一致。采用混合线性模型(Mixed linear model)对6个纤维品质性状的关联分析检测到65个关联位点(< 0.01), 各位点对表型变异贡献率为2.57%~8.12%。本研究旨在利用重测序数据开发全基因组范围的可用于凝胶检测的InDel标记, 为棉花种质资源研究和分子标记辅助选择育种提供便捷工具。
陆地棉; InDel标记; 遗传多样性; 群体结构; 关联分析
棉花作为世界上重要的经济作物, 是纺织制造业中天然纤维的主要来源。陆地棉()是棉花的主要栽培种。我国棉花育种史较短, 前期改良主要依赖于国外引种[1], 当前选育和种植的大部分陆地棉品种多由美国引进的岱字棉、斯字棉、金字棉等少数种质资源衍生而来, 这也导致我国陆地棉种质资源相对匮乏、遗传基础较为狭窄、多样性水平低[2]。为有效拓宽陆地棉的遗传基础、加快不同育种目标下的棉花新品种选育及遗传改良进程, 将传统育种方法和分子标记辅助选择育种方法结合是有效可行的。目前, 多种分子标记如简单序列重复(SSR)、单核苷酸多态性(SNP)等广泛地应用于棉花种质资源遗传多样性评价、亲缘关系鉴定、遗传图谱的构建、重要农艺性状QTL定位及图位克隆和种质鉴定等方面[3-4]。
InDel标记是根据不同个体基因组同源序列发生的核苷酸片段插入或缺失而开发的, 具有基因组内分布广、密度高、变异稳定、可重复性好、价格低廉等优点, 在水稻[5]、玉米[6]、油菜[7]等作物中被广泛应用。2015年Zhang等[8]完成了异源四倍体陆地棉品种‘TM-1’全基因组测序及组装工作。2017年Wang等[9]完成了352份棉花材料重测序, 同时发掘大量InDel变异位点, 为全基因组范围的InDel标记开发提供了条件。
棉花许多重要的农艺性状如产量、纤维品质、生育期等均为数量性状, 解析复杂数量性状的遗传基础主要有连锁分析和关联分析两种方法。关联分析是一种以连锁不平衡为基础, 鉴定自然群体目标性状与遗传标记关系的分析方法。它具有无需构建作图群体、省时省力、解析精度高等优点, 也是传统连锁分析QTL定位的有益补充。在棉花中, 国内外广泛开展了关联分析相关的研究工作[9-11], 然而这些研究大部分是基于SNP的, 利用InDel进行关联分析的研究相对较少。
本研究基于Wang等[9]的重测序结果, 筛选InDel变异位点, 开发覆盖全基因组范围的、可用于凝胶检测的InDel标记, 并对重测序的262份陆地棉材料进行遗传多样性、亲缘关系和群体结构的分析, 以及通过关联分析挖掘与纤维品质性状相关联的InDel标记, 从而评价这些标记在棉花遗传和育种中的应用潜力。
1 材料与方法
1.1 供试材料及性状表型数据
采用262份陆地棉种质资源材料, 根据地理来源分类, 包括中国长江流域棉区的75份、中国黄河流域棉区的118份、中国西北内陆棉区的33份、中国北方特早熟棉区的17份、中国南方棉区的2份, 以及美国的11份和前苏联的6份[9]。依据Nie等[11]的SSR标记在503份种质材料中的扩增结果, 挑选了24份多态性高的材料用于后续多态性标记筛选, 这些材料比较全面地覆盖了种质材料的多态性信息, 利用24份材料作为参考, 可以快速筛选出具有代表性的多态性标记。24份种质材料编号分别为ZY40、ZY43、ZY54、ZY91、ZY121、ZY122、ZY211、ZY238、ZY240、ZY262、ZY320、ZY358、ZY361、ZY362、ZY376、ZY410、ZY413、ZY423、ZY434、ZY452、ZY468、ZY481、ZY495和ZY507。
种质材料的表型数据在Nie等[11]的文章中已报道, 从中获取了2年4点共8个环境下的表型数据及最佳线性无偏预测(BLUP)后的表型值, 8个环境分别为2012年湖北黄冈(E1)、2012年河南原阳(E2)、2012年新疆库尔勒(E3)、2012年新疆石河子(E4)、2013年湖北黄冈(E5)、2013年河南原阳(E6)、2013年新疆库尔勒(E7)和2013年新疆石河子(E8)。选取与纤维品质相关的6个表型数据, 分别为纤维上半部平均长度(FUHML)、纤维比强度(FS)、纤维伸长率(FE)、纤维整齐度(FU)、马克隆值(MV)和短纤维率(SF)。
1.2 InDel标记设计
基于Wang等[9]检测的InDel位点集合, 筛选出测序深度大于7、插入/缺失碱基数大于30 bp且最小等位基因频率大于0.05的InDel位点。提取InDel位点上下游150 bp序列, 将其中含有“N”的位点过滤。以Zhang等[8]发表的陆地棉标准品系‘TM-1’基因组序列为参考进行比对, 阈值设置为1×10–10, 选取完全匹配的InDel位点作为候选标记开发位点。使用Primer 3.0[12]设计引物, 引物长度范围为18~24 bp, GC含量范围在40%~60%之间, 退火温度范围为54~60℃。最后, 去除位于scaffold上的引物及SSR引物。引物由金斯瑞生物科技有限公司合成。
1.3 基因组DNA的提取、PCR扩增及电泳检测
从田间取幼嫩叶片, 参照Li等[13]改良的CTAB法提取基因组DNA。以微量分光光度计Nanodrop 2000检测其质量和浓度, 并稀释至终浓度20~40 ngμL–1, 于–20℃保存备用。
采用10 μL PCR扩增体系, 其中包括1×reaction buffer、0.8 mmol L–1MgCl2、0.25 mmol L–1dNTPs、2.5 nmol L–1primers、25 ng DNA、0.5 UDNA polymerase (上海博彩生物科技有限公司)。扩增程序为95℃预变性5 min; 然后95℃变性50 s, 退火45 s (根据不同引物设置退火温度), 72℃延伸60 s, 34个循环; 最后72℃延伸5 min, 15℃保存。用6%非变性聚丙烯酰胺凝胶电泳分离扩增产物, 银染检测分离条带。
以人工读带, 同一位置上清晰且重复性好的条带记为“1”, 无带记为“0”, 缺失数据记为“–”。
1.4 数据统计分析
使用POWERMARKER V3.25软件[14]计算多态性信息含量(polymorphic information content, PIC)和基因多样性指数; 计算各种质材料间Nei’s遗传距离,依据距离矩阵构建系统发生树, 进行Neighbor- Joining聚类分析, iTOL[15]用于绘制和编辑聚类图。
利用STRUCTURE 2.3.4软件[16]计算各种质材料相应的Q值, 用于分析自然群体遗传结构。设置组群数值为1~10, 每个值独立重复运算7次, 将MCMC (Markov Chain Monte Carlo)开始时的不作数迭代(Length of burn-in period)设为100,000次, 再将不作数迭代后的MCMC设为100,000次; 利用STRUCTURE HARVESTER[17]分析输出数据, 依据ln()和Δ值来确定最佳K值[18]。
利用TASSEL 5.0软件[19]进行主成分分析(Principal component analysis, PCA), 估算PCA矩阵对表型变异的解释率。同时, 以群体结构(Q)和亲缘关系()为协变量的混合线性模型(MLM)进行性状-基因型的关联分析, 计算各标记位点在<0.01时对表型变异的贡献率(2)。
2 结果与分析
2.1 InDel标记开发及多态性分析
基于Wang等[9]重测序检测的InDel位点, 筛选并开发了3206个InDel标记, 可在网页(http://cotton. hzau.edu.cn/EN/download.php)中下载标记的相关信息, 文件名称为“ZY_InDels data”。在棉花基因组中平均每隔5.88 Mb挑选一个标记, 最终挑选出均匀分布于26条染色体的320个标记进行合成。
利用24份多态性高的种质材料进行多态性标记的初步筛选, 检测到87个多态性标记, 占总标记数的26.88%, 详细信息见附表1。多态性标记涵盖了整个棉花基因组, 每条染色体至少一个标记, 平均每条染色体上3.35个。图1为标记HAU_ID_ D11-01在部分材料中的扩增结果。其他230个标记无多态性, 3个标记没有得到扩增产物。

图1 标记HAU_ID_D11-01在部分品系中扩增结果
2.2 遗传多样性分析
利用87个多态性标记对262份棉花种质资源材料进行基因型分析, 共检测到160个等位变异位点, 平均每对标记1.84个。基因多样性指数变幅为0.0874 (HAU_ID_D07-04)~0.5000 (HAU_ID_D05- 08), 平均值为0.3876; 多态性信息含量(PIC)变幅为0.0836 (HAU_ID_D07-04)~0.3750 (HAU_ID_A07- 02), 平均值为0.3073 (附表1)。
2.3 群体结构分析
ln()值随假定亚群数值(1~10)的增大而持续增大。在=2时Δ达到最大(图2), 由此, 在遗传结构上将262份棉花种质材料划分为POP-1和POP-2两个亚群, 绘制供试材料的群体结构图(图3)。POP-1包含175份材料, 黄河流域棉区(84份)和长江流域棉区(71份)的种质材料占该组的88.57%; POP-2包含87份材料, 黄河流域棉区(34份)、西北内陆棉区(23份)和北方特早熟棉区(13份)的种质材料占该组的80.46%。每个亚群都包含不同种植区域的种质材料, 但同时又以同一种植区域的种质材料为主。
主成分分析(PCA)中PC1、PC2分别解释了个体变异的7.58%和4.55%。以PC1为x轴、PC2为y轴构建一个二维坐标图(图4)。种质材料大致上可分为2个集群, 一个以长江流域棉区和黄河流域棉区的种质材料为主, 另一个以黄河流域棉区、西北内陆棉区和北方特早熟棉区的种质材料为主。不同来源的材料在一定程度上相对独立, 而伴随育种过程中的基因交换和基因渗入, 材料互相混合。PCA观察结果与STRUCTURE软件的分析结果较为一致。
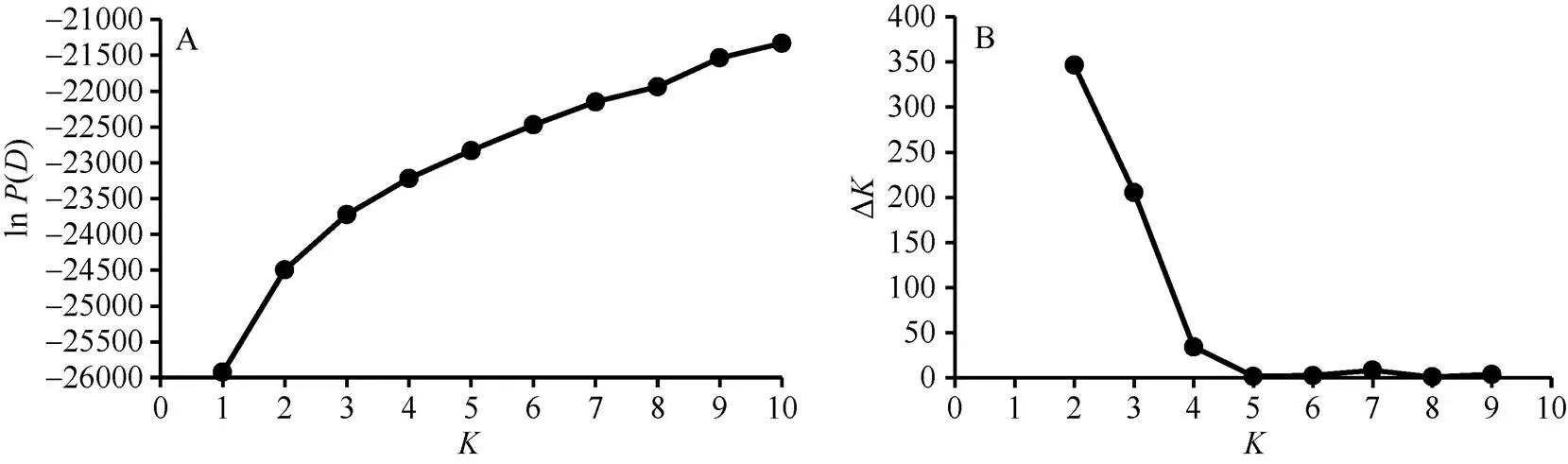
图2 K值与ln P(D)、ΔK值折线图
A: ln()与值变化折线图; B: Δ值随值变化折线图。
A: Line chart of ln() value with change of; B: Line chart of Δvalue with change of.
2.4 基于遗传距离的聚类分析
262份材料的Nei’s遗传距离(Genetic distance, GD)平均为0.499, 最高为1.075, 最低为0.012。遗传距离最高的材料均来自黄河流域棉区, 分别是ZY31 (晋棉13)和ZY297 (晋棉9)。基于距离矩阵构建系统发生树, Neighbor-Joining聚类图(图5)显示262份陆地棉材料可以划分为G1 (红色线条)和G2 (绿色线条)两大类群, G1包含182份种质材料(中国黄河流域棉区93份、中国长江流域棉区70份、中国西北内陆棉区7份、中国北方特早熟棉区5份、美国5份、前苏联2份); G2包含80份种质材料(中国黄河流域棉区25份、中国长江流域棉区5份、中国西北内陆棉区26份、中国北方特早熟棉区12份、中国南方棉区2份、美国6份、前苏联4份), 该组材料来源更为丰富, 其平均遗传距离也更高。总体来看, 262份种质材料的聚类分析结果与系谱信息相吻合。

图3 基于InDel标记的262份陆地棉品系群体结构图
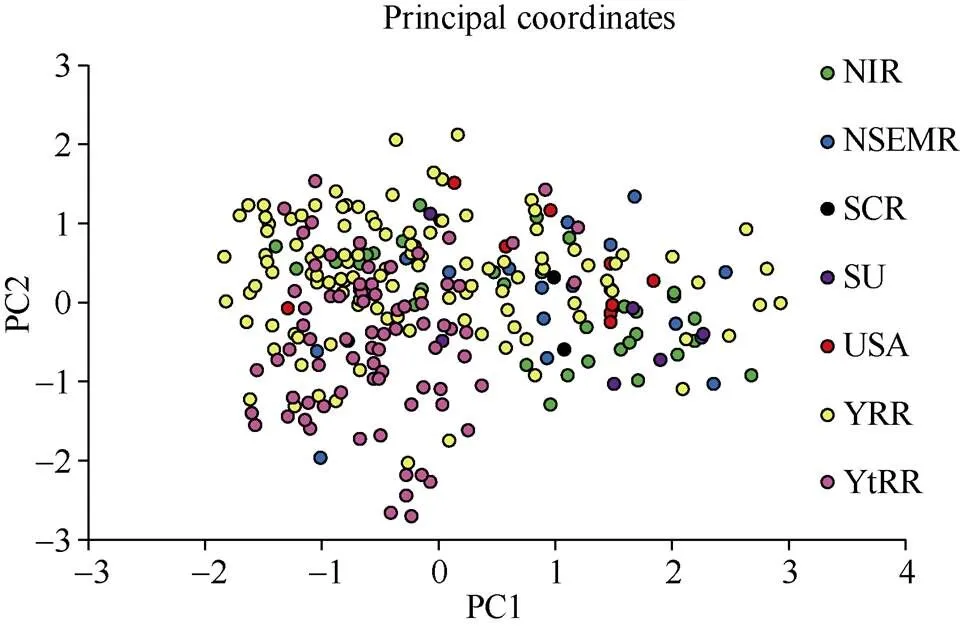
图4 基于InDel标记的262份陆地棉品系的PCA图
NIR: 中国西北内陆棉区; NSEMR: 中国北方特早熟棉区; SCR: 中国南方棉区; SU: 前苏联; USA: 美国; YRR: 中国黄河流域棉区; YtRR: 中国长江流域棉区。
NIR: Northwestern Inland Region of China; NSEMR: Northern Specific Early Maturation Region of China; SCR: South China Region; SU: Former Soviet Union; USA: American; YRR: Yellow River Region of China; YtRR: Yangtze River Region of China.
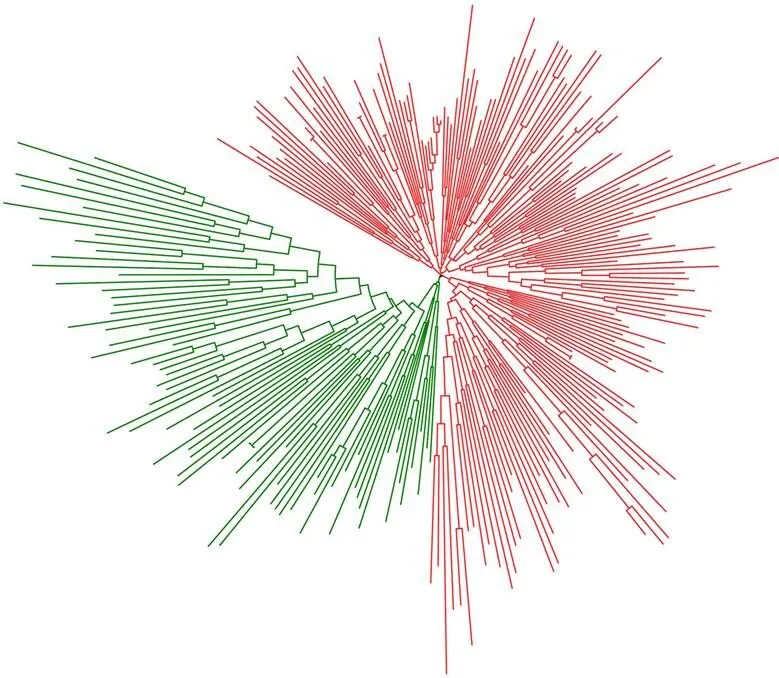
图5 基于遗传距离的系统发生树
红色线条: G1, 包含182份种质材料; 绿色线条: G2, 包含80份种质材料。
Red lines: group 1, including 182 accessions; Green lines: group 2, including 80 accessions.
2.5 性状-标记的关联分析
采用TASSEL 5.0软件中MLM(Q+K)模型进行性状-标记的关联分析。表型性状包括E1~E8和最佳线性无偏预测(BLUP)的表型值。在<0.01显著条件下, 共检测到65个与纤维品质性状相关联的位点(附表2)。与上半部纤维长度相关联的位点有12个, 在不同环境中对表型变异解释率范围为2.97% (HAU_ID_D12-10)~5.92% (HAU_ID_D09-10), 平均值为3.43%。对其余5个纤维品质性状, FS、FE、FU、MV和SF分别检测到7、13、10、9和14个关联位点,表型变异的解释率范围分别为2.57%~7.36%、2.61%~5.66%、2.74%~8.12%、2.72%~5.58%和2.66%~ 6.99%。65个关联位点中, 有23个至少能在2个环境中被检测到, 有11个至少能在4个环境中被检测到。其中, 标记HAU_ID_D02-06与FE在8个环境中均被检测到关联, 标记HAU_ID_D12-10与FU在6个环境中被检测到关联, 标记HAU_ID_A01-15与FL在5个环境中被检测到关联。
同一标记位点与多种性状相关联可能是多效性引起的。统计发现19个标记位点为多效应位点, 这些位点至少与2个性状相关联(表1)。其中, 标记HAU_ID_D07-09同时与5个性状(FUHML、FS、FE、FU、SF)相关联; 标记HAU_ID_D12-10同时与4个性状(FUHML、FS、FU、SF)相关联; 5个标记同时与3个性状相关联, 分别为HAU_ID_A01-15 (FUHML、FU和SF)、HAU_ID_A03-09 (FE、FU和SF)、HAU_ID_A10-09 (FS、FU和MV)、HAU_ID_ D06-06 (FUHML、FU和SF)和HAU_ID_D06-07 (FUHML、FU和SF); 其余12个位点与2个性状相关联。
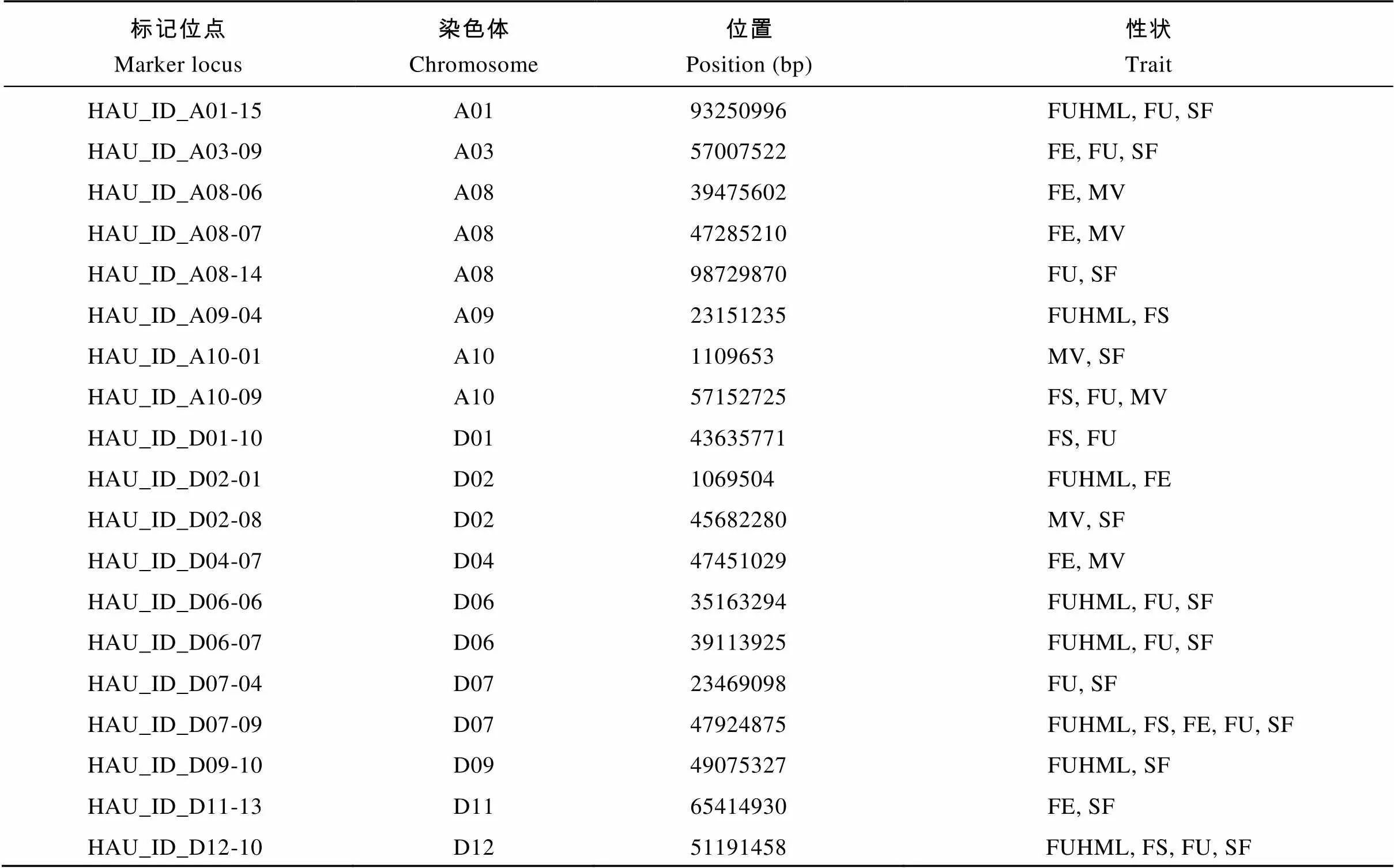
表1 多效应标记位点
3 讨论
3.1 InDel标记开发及应用
InDel变异广泛存在于基因组中, 其多态性频率仅次于SNP。InDel标记以其密度大、准确性高、具共显性、检测的经济快速和高效等特点成为分子标记开发的宝贵资源。2017年Wang等[9]对352份种质材料进行重测序, 我们挑选了其中262份陆地棉材料进行InDel标记开发, 由于中等大小的插入/缺失位点易于利用凝胶电泳进行基因分型, 参考Wu等[20]的InDel筛选标准, 筛选出插入/缺失片段长度大于30 bp的InDel位点, 并将之转化为基于PCR的可用于凝胶检测的InDel标记, 最终设计了3206个InDel标记并从中挑选了均匀分布于基因组的320个InDel标记用于多态性验证。
为了快速筛选多态性标记, 我们根据Nie等[11]的SSR标记在503份种质材料中的扩增结果, 挑选出具有代表性的、材料间多态性丰富的24份种质用于标记筛选。以320个InDel筛选到87个多态性标记, 多态性比例为26.88%。在陆地棉中, Wang等[21]基于RAD-seq测序数据开发了121对InDel标记, 两亲本间多态性率为25.62%, 与本研究结果相似, 均呈现出一个较低的多态性水平。从实验方法上来看, 本研究仅采用24份种质材料筛选多态性标记, 会导致部分标记的多态性不能被检测。同时, 采用聚丙烯酰胺凝胶电泳检测标记的多态性, 受凝胶的分辨率及分离效果等因素影响, 导致多态性标记的识别能力不够。此外, 陆地棉是异源四倍体物种, 基因组较为复杂, A亚组和D亚组间同源性程度较高, 在目前的基因组序列质量还不够完善的情况下, 重测序的序列同基因组序列比对时可能将来自不同亚组的序列比对到同一位置, 造成假阳性, 因而无法检测到标记的多态性。但与以往在陆地棉种内开发的SSR标记多态性率(2.5%~6.3%)相比[21-23], 基于重测序开发的InDel标记多态性率有所提高, 该研究结果为将来陆地棉引物的开发提供了参考。
目前, SSR标记及SNP标记在棉花的遗传资源评价、种质鉴定、遗传图谱构建和基因定位与图位克隆[22,24-25]等方面广泛应用, 而InDel标记则报道较少。新一代测序技术的发展和应用, 为棉花基因组测序及高密度InDel标记的开发提供了良好的条件[8-9],可以预见InDel标记将有广阔的应用前景。
3.2 遗传多样性分析
种质资源是作物遗传改良的基础, 由于陆地棉品种同质性较强, 遗传基础相对狭窄, 分析陆地棉种质资源材料的遗传多样性、群体结构及亲缘关系对拓宽棉花种质基础及棉花育种起到重要作用。
本研究筛选的87个InDel标记均匀分布于棉花基因组上。基因多样性指数分布在0.087,4到0.500,0之间; 多态性信息含量分布在0.083,6到0.387,6之间, 均值为0.307,3, 能够较为全面地分析陆地棉的遗传多样性。此前, Huang等[10]以63,058个SNPs对503份种质资源材料进行分析, PIC值变幅为0.279~0.369, 均值为0.332。Fang等[24]挑选448对SSR标记分析了193份地理来源不同的陆地棉材料, PIC均值为0.29, 根据资料考证, 他们指出自20世纪以来美国陆地棉品种的遗传多样性逐渐下降。Ai等[26]和Tyagi等[27]的结果也表明陆地棉遗传多样性水平普遍偏低。本研究与前人研究结果基本一致。
3.3 聚类分析和群体结构分析
基于InDel标记的聚类分析将262份种质材料大致划分为两类(图5)。聚类结果往往受到群体大小及群体内成员构成差异等因素的影响, 2017年Wang等[9]基于全基因组SNP标记将352份种质材料划分为两类, 能有效地将中国与国外的种质及野生种质区分开。本研究所采用的262份种质材料是从352份材料中挑选得来的陆地棉, 剔除了大量野生种质及国外种质, 仅与Wang等[9]的国内种质材料聚类模式相比, 聚类结果较为一致, 均能将多数西北内陆棉区、北方特早熟棉区的材料与其他材料区分开。同时, 我们发现G1组89.6%的材料来源于长江流域和黄河流域, 由于地理位置相近、基础种质资源相同或相似及育种过程中的人工定向选择, 使得这两大棉区的材料遗传距离相近, 遗传差异较小, 更加倾向于聚在一簇。G2组中不同地理来源的材料互相混合, 多数材料遗传距离较远。后续可考虑通过加强不同棉区的种质交流、加强国外引种力度来有效丰富我国棉花品种的遗传多样性。
对群体结构的正确评估是进行复杂性状关联分析的前提[28]。群体结构分析结果表明262份材料可大致分为2个亚群, 与聚类分析结果相对比, POP-1与G1、POP-2与G2共有的种质材料分别为161份和66份, 重叠率分别为88.46%和82.50%。本研究结合了STRUCTURE、聚类分析及主成分分析(PCA)这3种方法对群体结构进行分析, 结果一致性较好, 可有效避免由群体结构分层、等位基因分布不均等造成的标记与性状间的虚假关联。
3.4 关联分析
产量和纤维品质是陆地棉主要的育种目标性状, 多是复杂的数量性状, 受环境影响较大, 遗传基础复杂。近年来, 关联分析广泛应用到棉花各性状尤其是纤维品质性状QTL定位中, 并挖掘到很多与陆地棉纤维品质性状相关的QTL[4, 9-11]。不同的关联分析方法得到的结果不尽相同。以群体结构(Q)和亲缘关系(K)作为协变量的MLM (Q+K)优于GLM (Q)和MLM(K), 能有效降低关联的假阳性, 提高检测结果的准确度和真实性[29]。
本研究利用多环境的表型数据进行标记-性状的关联分析。36个标记共检测到65个关联位点(<0.01), 各位点对表型变异的解释率为2.57%~ 8.12%。标记的开发是基于Wang等[9]的重测序数据, 故参照其结果设置LD, 将置信区间设置为±296 kb。与前人的研究结果比较后发现8个位点已有报道, 其中4个位点与本研究关联到相同性状。其余28个位点可能是一些新的标记位点, 有待进一步的实验验证。
4个具有相同关联性状的标记中, 一个标记(HAU_ID_D05-02)与马克隆值相关, 且在5个环境中检测到关联, 该标记位点(Pos: D05_13953192)和Ma等[30]检测到的位点(Pos: D05_13941003)十分相近, 表型变异解释率达5.58%。其他3个标记(HAU_ ID_A07-02、HAU_ID_D02-01、HAU_ID_D04-07)都与纤维伸长率相关, 其中, 标记HAU_ID_A07-02在4个环境中检测到关联, 表型变异解释率为3.95%,该位点被Zhang等[31]和Fang等[32]共同检测; 标记HAU_ID_D02-01表型变异解释率为3.10%, 与Ma等[30]共同检测; 标记HAU_ID_D04-07在5个环境中检测到关联, 表型变异解释率达5.22%, 该关联位点在Wang等[9]、Huang等[10]以及其他两篇报道[33-34]中共同检测。2013年Liu等[35]鉴定到一个参与赤霉素信号响应的基因GhGASL, 组织表达模式分析表明该基因在棉花纤维中特异性表达, 可能参与纤维发育早期的纤维细胞的伸长, 将该基因的特异性引物比对到‘TM-1’基因组[8]上, 引物位点(Pos: D04_47893998)与本研究的标记位点HAU_ID_ D04-07 (Pos: D04_47451029)紧密连锁。
在不同报道中能够重复检测到的位点支撑了本研究的关联结果, 这些关联位点具有可重复性和稳定性, 可用于QTL精细定位中候选区间的确定, 同时有助于后期分子设计育种中材料的选择及鉴定。此外, 本研究检测到19个多效应位点同样值得关注, 例如位于D12染色体的标记HAU_ID_D12-10, 与之关联的4个性状(FUHML、FS、FU、SF)能在多个环境中稳定检测, 且标记对性状表型变异解释率处于相对较高的水平。这些多效应位点为解析不同性状之间的基因网络及遗传联系提供参考依据, 对指导育种中多性状协同改良具有重要价值。
4 结论
基于全基因组重测序数据设计了3206个InDel标记, 挑选320个标记进行合成并检测到87个多态性标记, 在陆地棉种内标记开发中呈现较高的多态性水平。262份陆地棉种质资源的遗传多样性分析表明种质材料的遗传基础较为狭窄。群体结构分析将种质材料大致划分为2个亚群, 与基于遗传距离的聚类分析及主成分分析的结果相同。关联分析检测到65个与纤维品质性状显著相关的标记位点, 其中23个标记位点能在多个环境中被重复稳定检测到。结果表明了基于重测序开发InDel标记是有效可行的, 本研究开发的标记为进一步解析陆地棉纤维品质的遗传机理提供了理论依据, 为棉花分子标记辅助育种提供了高效便捷的基因组工具。
附表 请见网络版: 1) 本刊网站http://zwxb. chinacrops.org/; 2)中国知网http://www.cnki.net/; 3) 万方数据http://c.wanfangdata.com.cn/Periodical-zuowxb. aspx。
[1] 喻树迅. 中国棉花产业百年发展历程. 农学学报, 2018, (1): 85–91. Yu S X. The development of cotton production in the recent hundred years of China., 2018, (1): 85–91 (in Chinese with English abstract).
[2] 董承光, 王娟, 周小凤, 马晓梅, 李生秀, 余渝, 李保成. 基于表型性状的陆地棉种质资源遗传多样性分析. 植物遗传资源学报, 2016, 17: 438–446. Dong C G, Wang J, Zhou X F, Ma X M, Li S X, Yu Y, Li B C. Evaluation on genetic diversity of cotton germplasm resources (L.) on morphological characters., 2016, 17: 438–446 (in Chinese with English abstract).
[3] Dong C G, Wang J, Yu Y, Li B C, Chen Q J. Association mapping and favourable QTL alleles for fibre quality traits in Upland cotton (L.).,2018, 97: e1–e12.
[4] Huang C, Shen C, Wen T W, Gao B, Zhu D, Li X, Ahmed M M, Li D, Lin Z X. SSR-based association mapping of fiber quality in upland cotton using an eight-way MAGIC population., 2018, 293: 793–805.
[5] Sahu P K, Mondal S, Sharma D, Vishwakarma G, Kumar V, Das B K. InDel marker based genetic differentiation and genetic diversity in traditional rice (L.) landraces of Chhattisgarh, India., 2017, 12: e0188864.
[6] Liu J, Qu J T, Yang C, Tang D G, Li J W, Lan H, Rong T Z. Development of genome-wide insertion and deletion markers for maize, based on next-generation sequencing data., 2015, 16: 601.
[7] Liu B, Wang Y, Zhai W, Deng J, Wang H, Cui Y, Cheng F, Wang X W, Wu J. Development of InDel markers forbased on whole-genome re-sequencing., 2013, 126: 231–239.
[8] Zhang T, Hu Y, Jiang W, Fang L, Guan X, Chen J, Zhang J, Saski C A, Scheffler B E, Stelly D M.Sequencing of allotetraploid cotton (L. acc. TM-1) provides a resource for fiber improvement.,2015, 33: 531–537.
[9] Wang M J, Tu L L, Min L, Lin Z X, Wang P C, Yang Q Y, Ye Z X, Shen C, Li J Y, Zhang X L. Asymmetric subgenome selection and cis-regulatory divergence during cotton domestication., 2017, 49: 579–587.
[10] Huang C, Nie X H, Shen C, You C Y, Li W, Zhao W X, Zhang X L, Lin Z X. Population structure and genetic basis of the agronomic traits of upland cotton in China revealed by a genome-wide association study using high-density SNPs., 2017, 15: 1374–1386.
[11] Nie X H, Huang C, You C Y, Li W, Zhao W X, Shen C, Zhang B B, Wang H T, Yan Z H, Dai B S, Wang M J, Zhang X L, Lin Z X. Genome-wide SSR-based association mapping for fiber quality in nation-wide upland cotton inbreed cultivars in China., 2016, 17: 352.
[12] Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers., 2000, 132: 365–386.
[13] Li X M, Yuan D J, Wang H T, Chen X M, Wang B, Lin Z X, Zhang X L. Increasing cotton genome coverage with polymorphic SSRs as revealed by SSCP., 2012, 55: 459–470.
[14] Botstein D, White R L, Skolnick M, Davis R W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms., 1980, 32: 314–331.
[15] Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees., 2016, 44: W242–W245.
[16] Pritchard J K, Stephens M, Donnelly P. Inferences of population structure using multilocus genotype data., 2000, 155: 945–959.
[17] Earl D A, Vonholdt B M. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method., 2012, 4: 359–361.
[18] Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study., 2010, 14: 2611–2620.
[19] Bradbury P J, Zhang Z, Kroon D E, Casstevens T M, Ramdoss Y, Buckler E S. TASSEL: software for association mapping of complex traits in diverse samples., 2007, 23: 2633–2635.
[20] Wu D H, Wu H P, Wang C S, Tseng H Y, Hwu K K. Genome-wide InDel marker system for application in rice breeding and mapping studies., 2013, 192: 131–143.
[21] Wang H, Jin X, Zhang B, Shen C, Lin Z. Enrichment of an intraspecific genetic map of upland cotton by developing markers using parental RAD sequencing., 2015, 22: 147–160.
[22] Lin Z X, Zhang Y X, Zhang X L, Guo X P. A high-density integrative linkage map for., 2009, 166: 35–45.
[23] Liu R Z, Wang B H, Guo W Z, Qin Y S, Wang L G, Zhang Y M, Zhang T Z. Quantitative trait loci mapping for yield and its components by using two immortalized populations of a heterotic hybrid inL., 2012, 29: 297–311.
[24] Fang D D, Li P, Thyssen G, Hinze L L, Percy R G. A microsatellite-based genome-wide analysis of genetic diversity and linkage disequilibrium in Upland cotton (L.)., 2013, 191: 391–401.
[25] Wen T W, Wu M, Shen C, Gao B, Zhu D, Zhang X L, You C Y, Lin Z X. Linkage and association mapping reveals the genetic basis of brown fibre ()., 2018, 16: 1654–1666.
[26] Ai X, Liang Y, Wang J, Zheng J, Gong Z, Guo J, Li X, Qu Y. Genetic diversity and structure of elite cotton germplasm (L.) using genome-wide SNP data., 2017, 145: 409–416.
[27] Tyagi P, Gore M A, Bowman D T, Campbell B T, Udall J A, Kuraparthy V. Genetic diversity and population structure in the US Upland cotton (L.)., 2014, 127: 283–295.
[28] Flint-Garcia S A, Thornsberry J M, Buckler E S. Structure of linkage disequilibrium in plants., 2003, 54: 357–374.
[29] Zhang Z W, Ersoz E, Lai C Q, Todhunter R J, Tiwari H K, Gore M A, Bradbury P J, Yu J M, Arnett D K, Ordovas J M, Buckler E S. Mixed linear model approach adapted for genome-wide association studies., 2010, 4: 355–360.
[30] Ma Z Y, He S P, Wang X F, Sun J L, Zhang Y, Zhang G Y, Wu L Q, Li Z K, Liu Z H, Sun G F, Du X M. Resequencing a core collection of upland cotton identifies genomic variation and loci influencing fiber quality and yield., 2018, 50: 803–813.
[31] Zhang S W, Feng L C, Xing L T, Yang B, Gao X, Zhu X F, Zhang T Z, Zhou B L. New QTLs for lint percentage and boll weight mined in introgression lines from two feral landraces intoacc TM-1., 2016, 135: 90–101.
[32] Fang L, Wang Q, Hu Y, Jia Y H, Chen J D, Liu B L, Zhang Z Y, Guan X Y, Chen S Q, Zhou B L, Du X M. Genomic analyses in cotton identify signatures of selection and loci associated with fiber quality and yield traits., 2017, 49: 1089–1098.
[33] Islam M S, Thyssen G N, Jenkins J N, Zeng L, Delhom C D, McCarty J C, Deng D D, Hinchliffe D J, Jones D C, Fang D D. A MAGIC population-based genome-wide association study reveals functional association ofgene with superior fiber quality in cotton., 2016, 17: 903.
[34] Sun Z W, Wang X F, Liu Z W, Gu Q S, Zhang Y, Li Z K, Ke H F, Yang J, Wu J H, Wu L Q, Zhang G Y, Zhang C Y, Ma Z Y. Genome-wide association study discovered genetic variation and candidate genes of fibre quality traits inL., 2017, 15: 982–996.
[35] Liu Z H, Zhu L, Shi H Y, Chen Y, Zhang J M, Zheng Y, Li X B. Cotton GASL genes encoding putative gibberellin- regulated proteins are involved in response to GA signaling in fiber development., 2013, 40: 4561–4570.
Development and evaluation of InDel markers in cotton based on whole-genome re-sequencing data
WU Mi, WANG Nian, SHEN Chao, HUANG Cong, WEN Tian-Wang, and LIN Zhong-Xu*
National Key Laboratory of Crop Genetic Improvement / College of Plant Science and Technology, Huazhong Agricultural University, Wuhan 430070, Hubei, China
Insertion and deletion (InDel) are abundant forms of genetic variation in the genome. InDel has been recognized as an ideal source for marker development due to its high-density distribution and genotyping efficiency. In this study, the whole genome re-sequencing data of 262 upland cotton accessions were applied to identify 3206 InDel markers, and 320 markers with uniform distribution across the genome were selected to be evaluated. Eighty-seven polymorphic markers were identified, accounting for 26.88% of screened markers. A total of 160 allelic loci were detected using the 87 polymorphic markers in the 262 upland cotton accessions with an average polymorphic information content (PIC) of 0.3073 (ranging from 0.0836 to 0.3750) and an average genetic diversity of 0.3876 (ranging from 0.0874 to 0.5000), indicating a relatively low genetic diversity. Population structure analysis revealed extensive admixture and identified two subgroups, clustering analysis and principal component analysis supported the subgroups identified by STRUCTURE. Association analysis were performed by MLM (Mixed linear model), and 65 marker loci were associated with fiber quality traits (< 0.01), explaining 2.57%–8.12% of the phenotypic variation. Genome-wide and gel based InDel markers developed based on re-sequencing data in this study provide a facile tool for cotton germplasm resources research and molecular marker assisted selection breeding.
Upland cotton; InDel marker; genetic diversity; population structure; association analysis
2018-07-19;
2018-10-08;
2018-11-16.
10.3724/SP.J.1006.2019.84100
林忠旭, E-mail: linzhongxu@mail.hzau.edu.cn
E-mail: 656542680@qq.com
本研究由湖北省技术创新专项(2018ABA082)资助。
This study was supported by the Technology Innovation Program of Hubei Province (2018ABA082).
URL: http://kns.cnki.net/kcms/detail/11.1809.S.20181115.1104.002.html
猜你喜欢
今日农业(2022年14期)2022-09-15
今日农业(2022年13期)2022-09-15
世界科学技术-中医药现代化(2022年3期)2022-08-22
江苏农业科学(2022年6期)2022-04-15
昆明医科大学学报(2022年2期)2022-03-29
中国棉花(2021年3期)2022-01-01
昆明医科大学学报(2021年6期)2021-07-31
昆明医科大学学报(2021年3期)2021-07-22
四川蚕业(2020年4期)2020-02-10
种子(2019年12期)2020-01-07
