
CDC25A与肿瘤的研究进展△
2019-01-15 08:28唐艳萍曹骥
癌症进展 2018年15期
唐艳萍,曹骥
广西医科大学附属肿瘤医院实验研究部,广西 南宁 530021
细胞分裂周期因子25(cell division cycle 25,CDC25)于1986年被Russell等[1]在裂殖酵母过程中分离出来,因其可以启动有丝分裂而得名。CDC25是一种双特异性磷酸酶,可以通过催化细胞周期依赖性蛋白激酶(cyclin-dependent protein kinases,CDK)分子中的抑制性磷酸化位点去磷酸化从而激活CDK,并调控应对DNA损伤,推动细胞周期运行。人类CDC25主要包括CDC25A、CDC25B、CDC25C 3种亚型,其中CDC25A是控制G1/S期和G2/M期检查点的“开关”蛋白,在维持DNA复制的稳定性和细胞分裂周期完整性上起到重要作用,已成为近年来的研究热点。CDC25A在多种恶性肿瘤中均呈过表达状态,而且CDC25A过表达与肿瘤患者的不良预后密切相关。因此,本文就CDC25A与肿瘤关系的研究进展作一综述。
1 CDC25A的概述
CDC25A的编码基因定位于染色体3p21上,编码酪氨酸磷酸酶。CDC25A蛋白由524个氨基酸残基组成,含有N端的调节结构域和C端的催化结构域。CDC25A的调节结构域含有多个磷酸化位点、核定位序列和核输出序列;磷酸化位点可以调节CDC25A的自身稳定性及与其他蛋白之间的相互作用;核定位序列和核输出序列可以调节CDC25A的亚细胞定位[2-3]。CDC25A的催化结构域含有一个HCX5R基序,相邻α螺旋的偶极矩有利于半胱氨酸去质子化,促进CDC25A与含有磷酸化苏氨酸和酪氨酸基团的底物结合[4]。CDC25A的活性受到了包括转录水平、翻译水平和翻译后修饰水平等多个层面的调控[5]。
2 CDC25A在肿瘤中的表达
目前,CDC25A被一致认为具有癌基因功能,在许多肿瘤细胞中存在过表达的现象,参与肿瘤的发生、发展过程,并与肿瘤的恶性程度和预后有关。Yamashita等[6]研究了人脑胶质瘤组织及正常脑组织中CDC25AmRNA的表达水平,结果显示脑胶质瘤组织中CDC25AmRNA的表达水平高于正常脑组织。Singh等[7]分析了CDC25A与视网膜母细胞瘤患者临床特征的关系,结果发现CDC25A与视网膜母细胞瘤的分化、转移有关。Brunetto等[8]选取313例乳腺癌患者为研究对象,通过免疫组织化学法检测发现,49.8%的乳腺癌患者的乳腺癌组织中存在CDC25A过表达;生存分析结果显示,CDC25A过表达与乳腺癌患者的总生存期和无病生存期缩短有关。Lu等[9]研究表明,CDC25AmRNA和蛋白在人肝癌组织中的表达水平均高于癌旁组织及正常组织,CDC25AmRNA在人肝癌组织中的表达水平与肝癌患者的临床分期、有无门脉瘤栓及肝外转移有关,但与肝癌患者的术后复发情况、肿瘤直径、病灶个数、血清甲胎蛋白水平和肿瘤分化程度无关。王红雷[10]通过免疫组织化学法检测CDC25A蛋白在肝癌组织中的表达情况,结果发现CDC25A蛋白的高表达与肝癌患者的病理分化程度、肿瘤大小及生存期有关。
3 CDC25A与肿瘤的发生发展
3.1 CDC25A与肿瘤细胞周期
细胞周期可分为DNA合成前期(G1期)、DNA合成期(S期)、DNA合成后期(G2期)及有丝分裂期(M期)4期,而G1/S期和G2/M期这两个阶段最为重要。CDC25A在G1/S期和G2/M期的进程中发挥着至关重要的作用。在G1/S期进程中,CDC25A蛋白可以促进抑制性磷酸残基脱磷酸化,激活CDK2和CDK4/6,而激活的CDK2和CDK4/6分别与相应的细胞周期蛋白cyclin E和cyclin D结合形成复合物,并通过复合物使肿瘤抑制基因Rb表达的蛋白磷酸化,从而使原作为E2F1转录抑制因子结合在E2F1上的Rb蛋白与E2F1分离,活化E2F1。活化的E2F1可以激活一系列下游事件,加快细胞由G1期进入S期,促进细胞增殖[5]。在G2/M期进程中,过表达的CDC25A与CDC25B、CDC25C协同作用于cyclin B1/CDK1,使CDK1的第14位苏氨酸和第15位酪氨酸去磷酸化[11]。同时,活化的cyclin B1/CDK1复合物通过磷酸化CDC25A的第17位色氨酸和第115位色氨酸,增加CDC25A的稳定性,因此cyclin B1/CDK1复合物和CDC25A形成了一个正反馈环通过G2/M期检查点。细胞周期调控异常是肿瘤发生的重要诱因之一,CDC25A作为细胞周期G1/S期和G2/M期进程的重要调控因子,其表达水平的增加会促进G1/S期和G2/M期检查点之间的转变,导致细胞生长失控从而引起恶性转化。
3.2 CDC25A与肿瘤细胞凋亡
细胞凋亡是一种由多基因严格控制的在一切生物发育过程中细胞自主有序的死亡途径。肿瘤的发生不仅与细胞的异常增殖和分化有关,还与细胞凋亡的调节紊乱存在密切关系。异常表达的CDC25A可以通过影响凋亡相关因子的生物活性丰度或定位,阻止细胞凋亡,诱导恶性肿瘤的发生。凋亡信号调节激酶1(apoptosis signal-regulating kinase 1,ASK1)是促分裂原活化的蛋白激酶激酶激酶(mitogen-activated protein kinase kinase kinase,MAP3K)家族成员之一,在调节细胞凋亡的过程中起着重要作用。ASK1可以激活p38丝裂原激活蛋白激酶(mitogen-activation protein kinase,MAPK)和JNK/SAPK通路,随后引起bcl-2、bcl-X、p53、caspase 3等细胞凋亡因子的活化,从而影响肿瘤细胞周期[12]。Zou等[13]运用酵母双杂交筛选系统和免疫共沉淀方法验证了CDC25A可与ASK1相互作用,并证实在不影响CDC25A磷酸化能力的前提下,CDC25A可以通过羧基端结合在ASK1激酶激活结合区域附近,抑制ASK1激酶活性,从而阻止细胞凋亡。核因子-κB(nuclear factor kappa B,NF-κB)信号转导途径可以通过多种方式抑制细胞凋亡,而CDC25A可以正向调控NF-κB,通过抑制细胞凋亡,促进肿瘤细胞存活。Hong等[14]在研究中发现,CDC25A可以通过增加核因子-κB抑制蛋白α(nuclear factor kappa B inhibitor alpha,Iκ-Bα)的第32位丝氨酸的磷酸化,降低Iκ-Bα蛋白的表达,随后引起NF-κB活性的增高;随后研究者将肿瘤细胞过表达CDC25A基因,同时抑制NF-κB活性或过表达Iκ-Bα,发现肿瘤细胞对顺铂的敏感性提高,肿瘤细胞凋亡率增加。此外,CDC25A对肿瘤细胞凋亡的调节与其在细胞质中的定位相关。Al-Matouq等[15]研究发现,CDC25A在人鳞状细胞癌中高表达,CDC25A从细胞核定位转移到细胞质定位,并与14-3-3蛋白相互作用,从而抑制肿瘤细胞凋亡。
3.3 CDC25A与DNA损伤应答
在细胞周期进程中,当DNA受到损伤时,细胞周期检测点便立即启动使细胞周期进程暂停。而CDC25A在此过程中扮演重要角色,通过调控CDC25A降解来阻滞细胞周期。DNA损伤使激活的ATM或ATR磷酸化下游的级联信号蛋白——细胞周期检验点蛋白Chk1和Chk2[16]。活化的Chk1可以磷酸化CDC25A的第82位丝氨酸,随后E3泛素连接酶SCF会结合到CDC25A的DSG基序上与β-TrCPl(F-box)一起促进CDC25A的泛素化降解,最终导致G1/S期阻滞[17-18]。此外,Chk1还可以通过磷酸化NEK11的第273位丝氨酸而激活NEK11,活化的NEK11通过磷酸化CDC25A的第82位丝氨酸,使CDC25A泛素化降解[19]。通过调节CDC25A的丰度,可以监控G2/M期转变,从而避免受损DNA进入细胞的有丝分裂进程[20]。活化的Chk2则通过磷酸化CDC25A的第123位丝氨酸,促进CDC25A降解,终止DNA合成并激活S期检验点[21]。CDC25A过表达时,发生DNA损伤的细胞无法在细胞周期检测点停止,细胞凋亡受到抑制,导致有基因缺陷的细胞癌变。
3.4 CDC25A与肿瘤细胞代谢
相关研究表明,CDC25A不仅可以参与细胞周期进程,还可以通过去磷酸化糖酵解关键酶——丙酮酸激酶 M2(pyruvate kinase M2,PKM2),参与肿瘤细胞的代谢调控[22]。在脑胶质瘤细胞中,表皮生长因子受体(epidermal growth factor receptor,EGFR)激活可导致c-src介导CDC25A的Y59位点磷酸化,激活的CDC25A使PKM2的S37位点去磷酸化,促进β-catenin反式激活和糖酵解基因GLUT1、PKM2及LDHA的表达,同时使CDC25A在正反馈环中上调自身表达。糖酵解基因的高表达可以明显提升细胞内糖酵解活力,促使肿瘤细胞进行大量的生物合成以保证细胞快速分裂、增殖。
3.5 CDC25A与肿瘤转移
Feng等[23]通过对CDC25A与乳腺癌患者临床特征的关系进行分析发现,CDC25A的表达与肿瘤转移有关。通过细胞和动物实验发现,过表达CDC25A可以提高乳腺癌细胞的转移能力,沉默CDC25A基因可以抑制裸鼠乳腺癌细胞的转移。进一步的机制研究证实,CDC25A可以通过去磷酸化CDK2增强FOXO1的稳定性,而FOXO1通过直接调控肿瘤转移相关因子基质金属蛋白酶1(matrix metalloproteinase,MMP1)的转录活性,促进肿瘤细胞转移。
4 CDC25A在肿瘤中过表达的调控机制
4.1 转录水平的调控
目前已知,有多种因子可以与CDC25A的启动子区域结合,正向或负向调控CDC25A的转录。E2F1通过与CDC25A基因启动子区域中的E2F-B区结合,调控CDC25A转录[24]。E2F1作为CDC25A的转录因子,在多种恶性肿瘤中呈过表达状态[25-26]。Barré等[27]研究证实,信号转导和转录激活因子3(signal transducer and activator of transcription 3,STAT3)依赖于MYC而调控CDC25A基因转录。采用白细胞介素-6处理血清饥饿48 h的肝癌细胞发现,STAT3可以与CDC25A基因的启动子区结合,启动CDC25A的转录;但当MYC基因被敲除之后,STAT3便不能激活CDC25A的转录。Yuan等[28]在以肝癌细胞HLE为对象的研究中,发现生物节律基因NPAS2可以通过直接结合CDC25A启动子nt-872到nt-866区域,促进CDC25A转录活化,进而使CDK2、CDK4和CDK6去磷酸化,促进肝癌细胞增殖及抑制肝癌细胞凋亡。人核酸酶敏感性结合蛋白-1(Y-box binding protein 1/nuclease-sensitive element-binding protein 1,YBX1)是另一种可调控CDC25A转录的因子。Zhao等[29]研究表明,在肺癌组织中,YBX1与CDC25A的表达呈正相关,随后CDC25A在肿瘤中过表达的调控机制较为复杂,可能归结于转录、转录后和翻译后的调控紊乱。(图1)该研究通过ChIP实验证实内源性YBX1结合在CDC25A启动子区域,过表达YBX1可以上调CDC25A启动子活性。
除了E2F1、STAT3等转录正向调控因子外,还存在CDC25A的转录负向调控因子。p53蛋白作为一种转录抑制因子,可以抑制CDC25A基因的转录。但CDC25A基因上并没有p53蛋白的结合位点。随后在以结肠癌细胞为研究对象的研究中发现,CDC25A的启动子序列上有一个ATF3的结合区域,因此推测ATF3可能通过介导p53,诱导CDC25A的转录抑制,从而维持肿瘤细胞周期阻滞[30]。此外,在低氧诱导的结肠癌细胞中发现,p21CiPl在miRNA-21的协同作用下,下调CDC25A的表达量[31],这可能与p21CiPl可以结合到CDC25A的启动子区,抑制CDC25A的转录有关。已有研究表明,肿瘤抑制因子SIRT6可以通过靶向CDC25A抑制结肠癌干细胞的增殖,实验观察发现SIRT6能直接结合到CDC25A的启动子区域,降低H3K9乙酰化水平,从而降低CDC25A的表达[32]。
4.2 转录后水平的调控
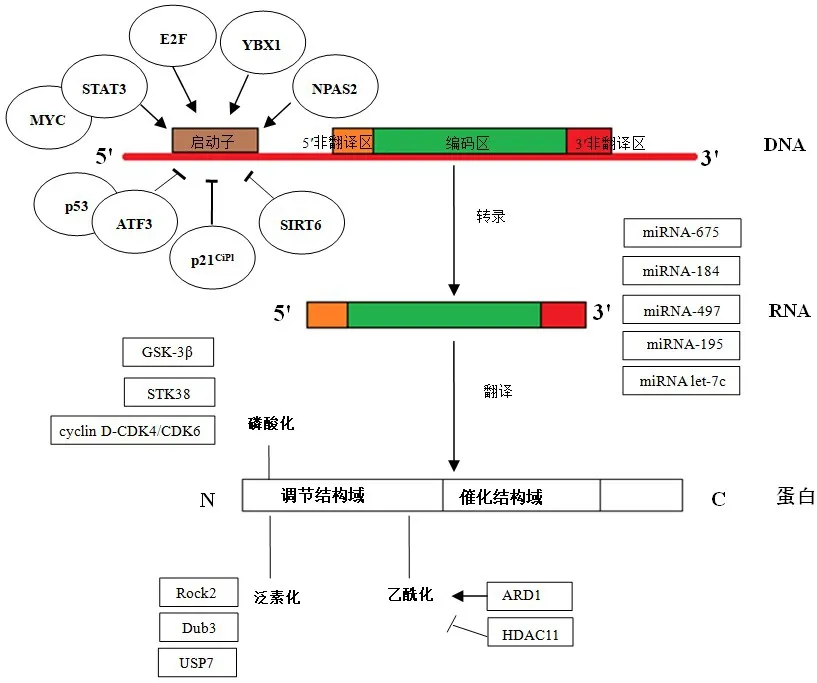
图1 CDC25A在肿瘤中过表达的调控机制
CDC25AmRNA的3'-UTR区含有多个miRNA的结合位点,miRNA与CDC25AmRNA的结合可促进CDC25AmRNA的降解或抑制CDC25A蛋白的翻译,从而调节CDC25A的水平。Furuta等[33]通过一系列筛选及表达验证,证明可抑制肝癌细胞增殖的miRNA-195和miRNA-497在肝癌细胞中表达下调,CDC25A为miRNA-195和miRNA-497的靶基因之一。Zhu等[34]研究发现,miRNA let-7c在肝癌细胞中表达量降低,运用荧光报告实验证实了CDC25A为miRNA let-7c的直接靶基因,miRNA let-7c可以通过与CDC25AmRNA的3'-UTR区结合,抑制CDC25A蛋白的表达。在MHCC-97H肝癌细胞中,miRNA-675可以正向调控CDC25A,促进细胞增殖,推测CDC25A在肝癌细胞中可能是miRNA-675 的靶基因[35]。Lin 等[36]研究表明,在非小细胞肺癌中miRNA-184的表达水平降低,其可与CDC25A的编码区结合,增加CDC25AmRNA的不稳定性,进而降低CDC25A的表达水平。
4.3 翻译后水平的调控
CDC25AmRNA被翻译后,会经历多种翻译后修饰,其中磷酸化、泛素化较为常见。Kang等[37]研究发现,GSK-3β是CDC25A的负性调控因子,PIK3可磷酸化CDC25A的第80位苏氨酸,进而引发GSK-3β磷酸化CDC25A的第76位丝氨酸,从而使CDC25A降解。Fukasawa等[38]研究证实,STK38直接磷酸化CDC25A的第76位丝氨酸,使CDC25A降解,导致G2/M检查点活化。相关研究表明,cyclin D-CDK4/CDK6复合物在G1期可磷酸化CDC25A的第40位丝氨酸,从而调控CDC25A的稳定性[39]。人体内CDC25A蛋白的稳定性还通过泛素依赖的蛋白酶体降解途径加以调控。Liu等[40]研究证实,肝癌细胞中高表达的Rock2可以与CDC25A直接相互作用,阻止CDC25A泛素化。有研究发现,乳腺癌细胞中去泛素化酶Dub3和CDC25A存在高表达现象,进一步分析证实,Dub3可以去除CDC25A的泛素链,使CDC25A半衰期异常延长和稳定性增加,导致CDC25A水平升高[41]。Biswas等[42]发现了与CDC25A有关的另一种去泛素化酶USP7,该研究以MCF7乳腺癌细胞为对象,发现沉默BRE的MCF7细胞的CDC25A蛋白表达降低;进一步研究发现,BRE通过募集USP7与CDC25A相互作用,调控CDC25A蛋白的表达。除了磷酸化和泛素化,目前还证实了乙酰化也可调控CDC25A的稳定性[43]。乙酰转移酶ARD1可直接与CDC25A结合,乙酰化CDC25A,使其半衰期延长,而HDAC11为CDC25A的脱乙酰酶。细胞受到DNA损伤后,乙酰化的CDC25A增加,并能调控其磷酸化活性,影响细胞周期,因此CDC25A、ARD1和HDAC11在多种肿瘤中存在异常表达。
5 CDC25A抑制剂
基于CDC25A在细胞周期、细胞凋亡及细胞代谢等过程中的关键作用,如能抑制该蛋白活性,则可控制肿瘤的发生和发展。针对CDC25A这一肿瘤治疗的潜在靶点,越来越多的CDC25A抑制剂陆续出现。
维生素K3及其先导的类似物是较早发现的CDC25抑制剂,它通过与CDC25催化域结合,导致形成无活性高度磷酸化的CDK1,抑制细胞周期,但不具有CDC25A、CDC25B、CDC25C特异性,且容易诱导产生活性氧。Rostom等[44]在合成的29种新1,2,4-三唑类化合物中发现,衍生物12、15、18、19和22号具有CDC25A、CDC25B磷酸酶特异性抑制性,在体外显示出较强的抗肿瘤作用,有作为CDC25抑制剂的潜在应用价值。Shen等[45]研究发现,环匹罗司在不影响CDC25AmRNA表达水平的情况下,可通过磷酸化CDC25A的第76位丝氨酸和第82位丝氨酸,诱导CDC25A蛋白降解,从而将乳腺癌细胞阻滞于G1期,抑制细胞增殖。近年来研究人员以苯醌为基本药效基团进行结构修饰,制备出许多新CDC25A抑制剂。Ge等[46]利用多药效模型筛选出具有特异性抑制CDC25A活性的化合物KM10389,证实其能显著抑制宫颈癌HeLa增殖,而对正常细胞无毒性作用。
除了化学药物,从很多的天然植物中提取的活性成分也被发现能调控肿瘤细胞中CDC25A的表达。Wang等[47]将青蒿琥酯用于胃腺癌细胞的实验中,证实青蒿琥酯可以抑制SGC-7901细胞的增殖及诱导细胞凋亡,其机制可能与青蒿琥酯调控CDC25A、bcl-2、BAX及caspase 3等有关。M22是从琉球松茎等其他药用植物和果实中分离出的羽扇豆醇衍生物,Yuan等[48]研究发现,浓度为6.80 μmol/L的M22可以通过抑制CDC25A、cyclin D1、cyclin E1和CDK2的表达,诱导非小细胞肺癌细胞A549阻滞在G1期,继而抑制细胞增殖。从小型坚果如蓝莓和葡萄等中提取的天然化合物紫檀芪,被证实可通过调控CDC25A、cyclin A2和CDK2的表达水平,抑制白血病细胞增殖及诱导细胞凋亡[49]。
6 小结
目前的研究提示,在肿瘤中异常表达的CDC25A表现出的原癌基因特性以及其在肿瘤细胞周期调控网络中的枢纽地位,使其逐渐成为抗癌药物研发中一个极具价值的分子靶标。鉴于CDC25A过表达分子机制的逐渐揭示,新结构、高活性、低毒性的CDC25A抑制剂不断涌新。然而CDC25A与其分子抑制物相互作用的规律尚未完全清楚,以及CDC25A活性位点的高敏感性给CDC25A抑制剂的研发及应用带来的难度等原因,目前尚未见其应用于临床治疗或预后方面的相关文献报道。因此,作为有前景的抗肿瘤药物靶点,CDC25A及针对其不同层面的调控机制研发的小分子靶向抑制剂还有待深入研究。
猜你喜欢
食品安全导刊(2022年17期)2022-07-04
中国农业科学(2022年12期)2022-06-28
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
中国畜牧杂志(2022年1期)2022-01-20
南昌大学学报(医学版)(2021年1期)2021-11-29
皮肤性病诊疗学杂志(2021年5期)2021-11-27
三农资讯半月报(2021年1期)2021-01-27
分析化学(2017年12期)2017-12-25
中国医药导报(2016年33期)2017-03-06
