
基于功率谱的蛋白质序列特征提取新方法
2019-01-11 02:13梁启浩唐旭清
食品与生物技术学报 2018年11期
梁启浩, 李 阳, 唐旭清
(江南大学 理学院,江苏 无锡 214122)
蛋白质序列特征提取是指依据研究的目的提取序列信息,并使用数学方法描述,建立可以反映序列结构和空间信息的特征向量,进而表达其功能[1]。如何从复杂的序列中挖掘有用的信息是生物信息学的研究方向之一,信号频谱分析技术基于自动信息处理,广泛应用于特征提取的各个领域,比如周期性分析、蛋白质编码区预测和基因识别等方面[2-3]。Yin等[2]将信号处理与分析方法引入DNA序列相似性分析中。Hota等[4]基于快速离散傅里叶变换(Fast discrete Fourier transform,DFT)和小波变换(Wavelet transform,WT),从功率谱等信号处理方法的角度对基因识别进行了研究。王其强等[5]基于功率谱将信号处理与分析方法应用于P53家族基因的三周期性特征分析。这些研究对于大数据中DNA序列处理过程中的特征提取有重要的意义。
蛋白质存在于所有的生物细胞中,是生命的物质基础之一,蛋白质序列的研究具有极其重要的意义。蛋白质空间结构的所有信息均隐藏在氨基酸序列中,因此研究蛋白质的氨基酸序列组成已经成为生物信息学研究领域的关键问题之一[6]。聚类分析技术已广泛应用于蛋白质序列信息处理的各个方面,如分析蛋白质间的亲缘关系,提取蛋白质结构信息、功能信息等[7-8],其目的是简约数据信息系统、降低系统复杂度。文献[9]通过Voss映射将DNA序列转换为数字序列,采用功率谱方法提取DNA序列的特征信息从而进行DNA序列聚类分析,其中特征信息提取的核心是由离散傅里叶变换的序列特征频谱的j(j=1,2,3)阶矩构造的一个12维的特征向量,并采用传统的非加权组平均法(UPGMA)得到不同物种基于这种相似关系的系统发生树。在此基础上,本文结合基于信号频谱分析技术与层次聚类方法,将DNA序列数据推广到蛋白质序列数据,进行蛋白质序列的特征提取与物种的系统发生树(或分层结构)研究。
1 材料与方法
1.1 数据来源
本文从NCBI网站中下载了文献[10]中19种动物 的 ND1、ND4 的 蛋 白 质 序 列 (NADH dehydrogenase subunit1是线粒体NADH脱氢酶亚基1的简写、NADH dehydrogenase subnits4是线粒体NADH脱氢酶亚基4的简写,分别表示为数据1与数据2)进行研究,具体的数据有Gibbon(NC_002082.1),Gorilla(NC_011120.1),Human(NC_012920.1),Chimp(NC_001643.1),Pygmy Chimp(NC_001644.1),Sumatran Orang (NC_002083.1),Bornean Orang(NC_001646.1),Hedgehog(NC_002080.2),Rat(AC_000022.2),Mouse(NC_005089.1),Donkey(NC_001788.1),Horse(NC_001640.1),Cow (NC_006853.1),Baleen whale (NC_001601.1),Fin whale (NC_001321.1),Cat(NC_001700.1),Grayseal(NC_001602.1),Harbor seal (NC_001325.1),Rhino(NC_001779.1)。同时下载了文献[11]中11种β珠蛋白蛋白质序列(表示为数据 3), 分别为 Human(AAA16334),Gorilla(CAA43421),Chimpanzee(CAA26204),Lemur(AAA 36822),Rabbit(CAA24251),Goat(AAA30913),Bovine(CAA25111),Mouse(CAA24101),Rat(CAA29887),Opossum (AAA30976),Gallus(CAA23700) 进 行 研究。并同时找到3种数据所对应的DNA序列与文献[9]做比较。
1.2 符号序列的数字表达HP模型
随着生物信息学的发展,对于DNA序列中碱基进行数值化的映射有很多,如Voss映射、实数映射、Z-curve 映射及朱平等[12]建立的映射 φ∶GF(73)→C343等。
本文考虑氨基酸的物理和化学性质,在详细的HP模型中将20种氨基酸分成4大类,分别为极性亲水性(PQ),极性疏水性(PR),非极性亲水性(SQ)和非极性疏水性 (SR), 且 PQ={G},PR={A,V,L,I,P,F},SQ={S,T,C,N,Q,K,R,H,D,E},SR={W,M,Y}, 由此就将一条给定长度的蛋白质序列转化为一条由4类氨基酸构成的4元序列。
对含有n个氨基酸的蛋白质序列s=s1s2…sn,其中为组成此蛋白质序列的氨基酸,进行数据化定义
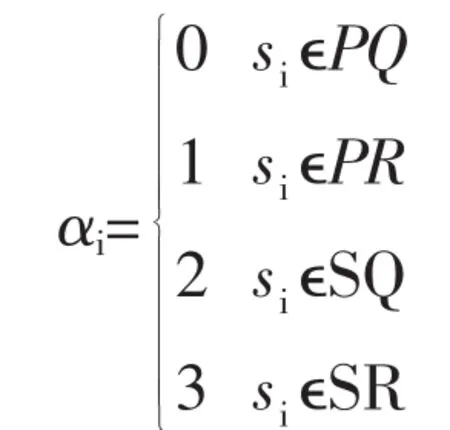
由此即可将任意一条蛋白质序列转化为一条由 0、1、2、3 构成的 4 元序列,记作:X(s)=α1α2…αn。这样产生的序列为基因序列的指示序列。
1.3 基于功率谱的蛋白质特征向量提取
里叶变换能将满足一定条件的某个函数表示成三角函数(正弦和/或余弦函数)或者它们的积分的线性组合。使用离散傅里叶变换,其显著的优点是使隐藏或潜伏在原始数据中的信息经周期性变换之后变得清晰。下文的研究以极性亲水性(PQ)为例说明,极性疏水性(PR),非极性亲水性(SQ)和非极性疏水性(SR)相似。
利用离散的傅里叶变换及上述的指示序列,可以将基因序列数据进行离散化
这样就能得到复数序列{UPQ(k)},k=0,1,…,N-1。对这个复数序列取模的平方,以定义序列的功率谱
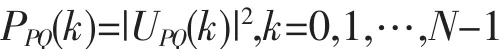
类似地,可得 PPR(k)、PSR(k)和 PSQ(k),且原氨基酸序列的功率谱为这4个子序列的功率谱之和。即
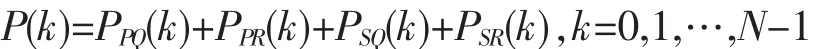
生物序列数学表示的目的之一就是分析生物序列的相似性[13],但是不同的蛋白质序列长度不同,因此仅通过功率谱不能进行相似性分析,进而去聚类。解决这种问题需要通过功率谱构造向量,而相似性则可以通过计算两向量之间的欧式距离得到。一般认为距离越小,两序列就越相似。对于极性且亲水性(PQ)类氨基酸,在文献[9]中定义j阶矩为
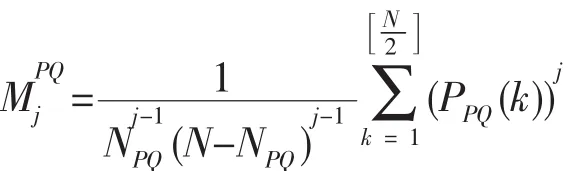
可以构建12维向量,也称为基于矩的
这样每一条蛋白质序列都会得到一个12维的特征向量。特征向量间的距离按欧式距离进行计算,即给定蛋白质序列i和j的特征向量分别为Mi和Mj,则两条序列之间的欧氏距离记为
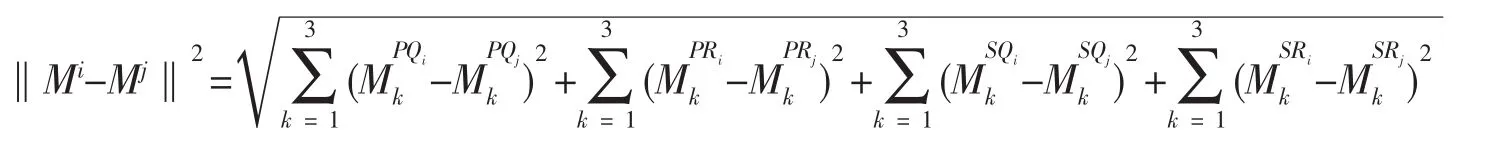
以下将在上面特征向量的基础上开展研究。
1.4 聚类
聚类分析是进行数据分析的一个基本方法,在数据挖掘、模式识别、生物信息学和统计学等领域都有广泛的研究与应用[14]。它是探索或提取隐含在数据中的新规律和新知识的重要手段。本文将采用基于文献[15]得到的层次聚类方法,在有限集X={x1,x2,…,xn}上定义一个标准化的度量矩阵d,令
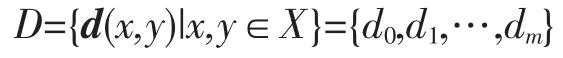
其中 d0=0<d1<…<dm。其算法如下:
算法A
S1 输入n个样本,i⇐0;
S2 构造n个类,每个类中只含有一个样本,记为 X(di)=C={c1,c2,…,cn};
S3 A⇐C,i⇐i+1,C⇐Ø;
S4 B⇐Ø;
S5 对于任意的 cj∈A,令 B⇐B∪{cj},A⇐A/cj;
S6 ∀ck∈A,如果存在 xj∈cj,yk∈ck,使得 d(xj,xk)≤di,则 B⇐B∪{cj},A⇐A/ck;
S7 C⇐{B}∪C;
S8 若A≠φ,则转S4;
S9 X(di)=C,输出 X(di);
S10 直到C≠{X},否则转S3;
S11 结束。
通过算法A,可获得数据系统的分层(或层次)结构。
1.5 聚类结果评价
本文采用熵作为聚类效果的度量标准,熵值表示同一类对象在聚类簇集中的分散程度。处于同一类中的对象熵值越高越分散,相应的聚类效果就越差。用标准公式计算簇i的熵,其计算公式为
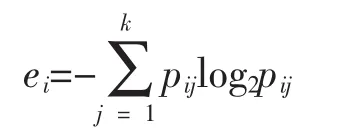
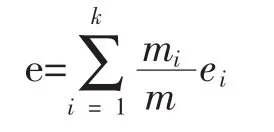
其中,k为簇的个数,m为数据集中对象的总数。熵值的含义是:熵值越小相应的聚类效果就越好。在理想情况下,每一类的所有对象应分布于不同的簇中,此时e=0。
2 实验结果比较与分析
将本文的方法用于ND1、ND4与β珠蛋白序列的相似性分析。选取这3种蛋白质序列来检验本文方法的好坏,一是因为这3种数据在生物上具有重要意义,研究的结果有实际应用价值,ND1与ND4参与线粒体氧化磷酸化的电子传递[10],β珠蛋白是位于红细胞内的一种更大的蛋白质 (血红蛋白)的一个组件(亚基)[11],对所有的生物体都是至关重要的。二是由于这3种数据已被广泛研究。将本文结果与文献[9]构建的结果进行比较,使本文的方法更具有说服力。
本文将数据分为3类时两种方法的错分率和熵值进行比较研究。图1~3为采用3种数据构造12维特征向量进行聚类分3类时的分层结构。表1,2是采用本文方法与文献[9]中的方法对实验数据进行聚类所得到的结果的对错统计情况,表3为采用两种方法将3种数据分3类时的熵值对比。
将数据1分为3类时,文献[10]中标准的分为3类时的层次结构为(Hedgehog,Donkey,Horse,Cow,Baleen whale,Fin whale,Cat,Gray seal,Harbor seal,Rhino), (Rat,Mouse), (Gorilla,Gibbon,Human,Chimp,Pygmy Chimp,Sumatran Orang,Bornean Orang),从图1和2可以看出,将本文的方法应用到数据1和数据2所得到的结果与文献[10]中的结果是基本一致。人(Human),苏门答腊猩猩(Sumatran Orang), 婆 罗 洲 猩 猩 (Bornean Orang), 长 臂 猿(Gibbon), 大猩猩 (Gorilla), 侏儒黑猩猩(Pygmy Chimp),黑猩猩(Chimp)彼此之间的线粒体 NADH脱氢酶很相似不是一种偶然,都属于灵长目。与其他哺乳类的动物进化关系最远的物种Mouse和Rat的线粒体NADH脱氢酶与其他物种最不相似,为啮齿目。19种数据中除(Rat,Mouse)属于脊索动物门以外,Hedgehog与另外16种都属于脊椎动物门,所以本文将数据中的Hedgehog先与灵长目聚为一类是可行的。而文献[9]的方法将数据1中本该属于鲸目、偶蹄目的Cow与啮齿目Mouse聚为1类,数据2中文献[9]的方法将啮齿目Rat和其他两类混在一起,而Mouse单独分为1类,这与进化事实相悖。因此,采用本文的方法得到的结果更好一些。
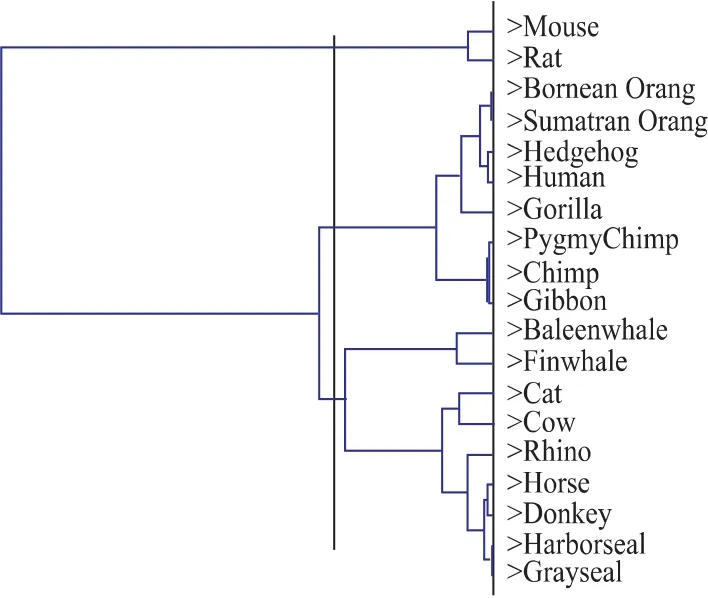
图1 数据1分3类时的分层结构Fig.1 Hierarchical structure of Data 1
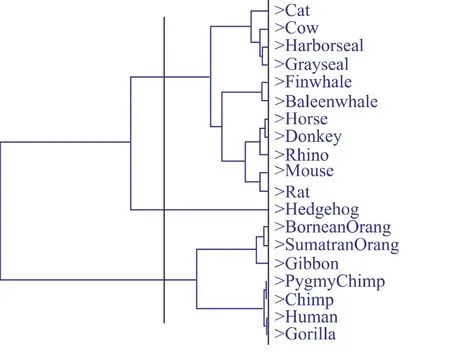
图2 数据2分3类时的分层结构Fig.2 Hierarchical structure of Data 2
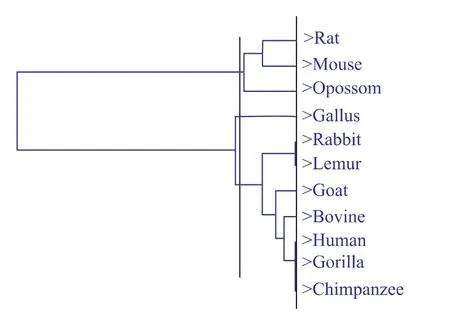
图3 数据3分3类时的分层结构Fig.3 Hierarchical structure of Data 3
将数据3分为3类时,文献[11]中标准的分为3类时的结果为(Human,Gorilla,Chimpanzee,Lemur,Rabbit,Goat,Bovine,Mouse,Rat), (Opossum),(Gallus)。从图3可以看出,将本文所得结果与文献[11]中的结果相比: 哺乳动物中的 (Mouse,Rat),(Human,Gorilla,Chimpanzee)和(Lemur,Rabbit)分别是聚类过程中距离最近的物种,它们处于同一分支上,即亲缘关系最近;Gallus作为11个物种中唯一的非哺乳动物和其他的哺乳动物之间的进化距离很远;本文将(Mouse,Rat)先与负鼠目Opossum聚为一类,文献[11]后将(Mouse,Rat)与 Opossum 聚为一类,有一定的差别。而采用文献[9]方法的聚类过程中,将Chimpanzee与Gorilla这两个物种单独分为一类,此与文献[11]差别较大,同时与进化事实不一致。因此,采用本文的方法研究β珠蛋白的结果好于文献[9]的方法。
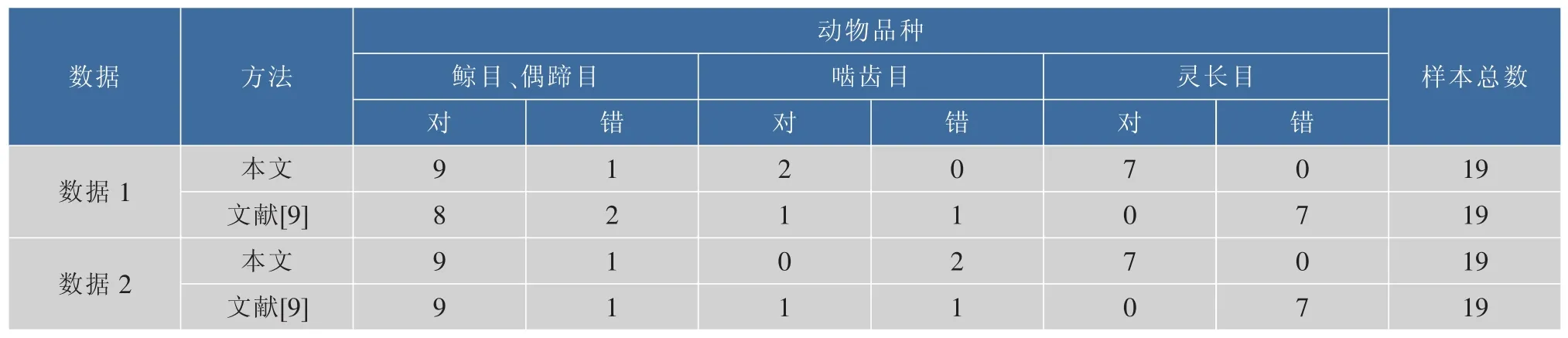
表1 数据1和数据2分3类时聚类结果对错统计Table 1 Error statistics of Clustering result for Data 1 and Data 2
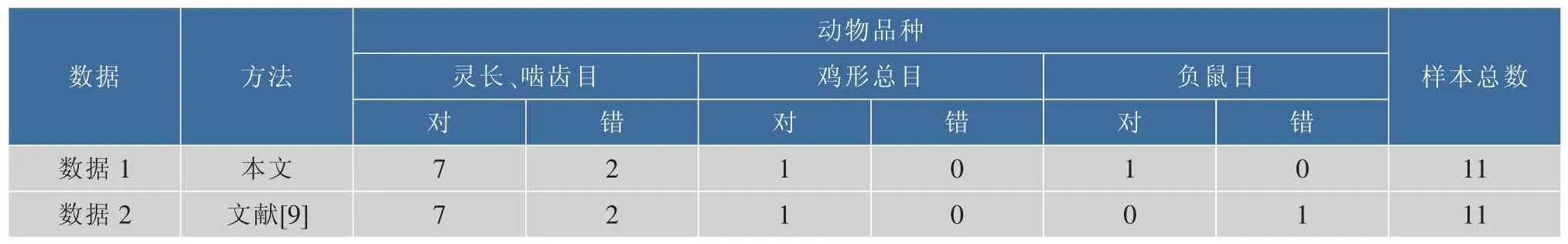
表2 数据3分3类时聚类结果对错统计Table 2 Error statistics of Clustering result for Data 3
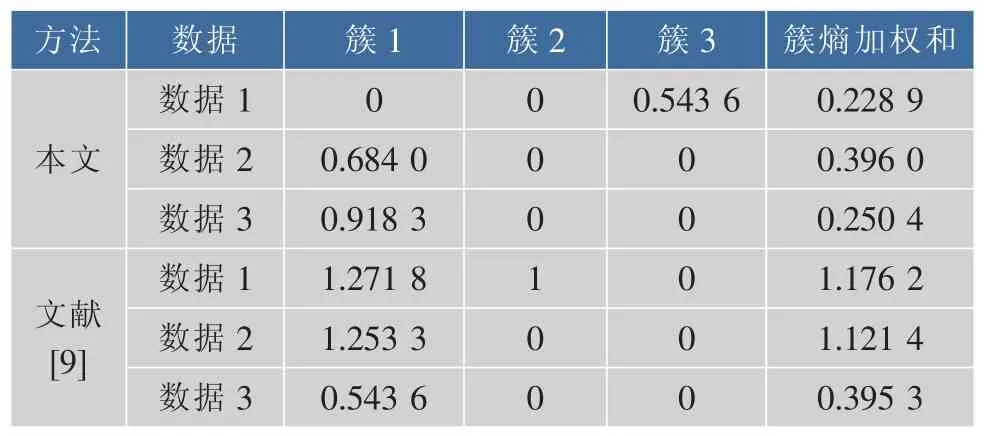
表3 本文方法与文献[9]的3类数据熵值比较Table 3 Compare the entropy values of our method with literature[9]on 3 Data sets
由表1,2可以看出将3种数据分别分为3类时,采用本文方法样本被分错类的数目分别为1个、3个、2个,错分率分别为 1/19=5.3%、3/19=15.8%、2/19=10.5%。与采用文献[9]的方法相比(样本被分错类的数目分别为10个、9个、3个,错分率分别为10/19=52.6%、9/19=47.4%、3/19=15.8%), 准确率分别提高了47.3%、31.6%、5.3%。可见将本文的方法应用于这3种数据的特征提取时能够提高聚类质量。
由表3可看出将3种数据分别分为3类时,采用本文方法簇熵加权和分别为0.228 9、0.396 0、0.250 4。与采用文献[9]的方法相比(簇熵加权和分别为 1.176 2、1.121 4、0.395 3),用本文方法产生的结果簇集具有最小的熵值,说明本文方法聚类结果较优。
因此,对于经常用于蛋白质氨基酸序列分析的小批量数据,由图1~3与表1~3可以看出用本文方法计算出的错分率与熵值都远低于文献[9]中的方法,即频率域上蛋白质序列的特征提取得到的相似度远远大于基于DNA序列的特征提取的相似度,本文的方法是有效的,之所以在蛋白质序列水平上得到的层次结构要比DNA序列更好,是因为对蛋白质序列的相似性进行分析,本质上是对组成蛋白质氨基酸序列的差异性比较,同一种蛋白质DNA序列比氨基酸序列长3倍,随着序列长度的增加,计算越来越复杂,并且序列到数字的映射以及对特征信息处理过程的信息缺失都会影响方法的准确率[2]。
通过本文方法来提取蛋白质序列的特征信息有3个优点:一是不用直接比较蛋白质序列而是去考虑经过离散傅里叶变换后这些蛋白质序列所对应的12维特征向量,特征向量是从组成蛋白质序列的氨基酸中提取出来的信息,这样蛋白质序列的比较就转化成了向量之间的比较;二是因为氨基酸序列的亲疏水特性、极性非极性跟蛋白质的结构有一定的关系,所以基于氨基酸的这一特性对其分类进而简化蛋白质序列,再提取特征向量可以包含了更多的生物信息;三是基于层次聚类构建的分层结构能非常直观地反映蛋白质之间的进化关系。尽管如此,在提取序列的特征向量时仍然会不可避免地伴随着某些蛋白质序列结构方面的信息丢失。这也正是目前在蛋白质的研究中面临的一大挑战。
3 结语
本文将基于DNA序列在频率域上的表示和离散傅里叶变换构造特征向量方法相结合以表征物种的特征方法进行推广,提出基于功率谱的蛋白质序列特征提取新方法。在研究的过程中采用经典HP模型对蛋白质的氨基酸序列数值化;由离散傅里叶变换将序列离散化,根据定义计算序列的功率谱构造蛋白质序列的特征向量距离;采用基于距离的层次聚类算法获取分层结构以考察蛋白质序列的相似性。选取3种不同的物种数据进行了新方法实验,以及与文献[9]中方法的比较研究。实验结果表明新方法是可行、有效的,且基于蛋白质的氨基酸序列在频率域上的特征提取方法优于基于DNA序列。这些研究结果对确定未知基因的结构与功能有重要的生物意义,对大规模的生物数据的自动信息处理具有应用价值。
猜你喜欢
九江职业技术学院学报(2022年1期)2022-12-02
保定学院学报(2022年2期)2022-04-07
时代英语·高一(2019年5期)2019-09-03
电子制作(2019年15期)2019-08-27
电子制作(2018年19期)2018-11-14
许昌学院学报(2018年4期)2018-05-02
中华建设(2017年1期)2017-06-07
自动化学报(2017年11期)2017-04-04
天然产物研究与开发(2016年1期)2016-06-05
电测与仪表(2016年11期)2016-04-11
