
儿童小脑萎缩的影像学诊断路径:111例患者回顾分析
2019-01-07 10:52肖江喜
中国临床医学影像杂志 2018年6期
赵 飞,肖江喜,谢 晟
(1.航天中心医院影像科,北京 100049;2.北京大学第一医院,北京 100034;3.中日友好医院,北京 100029)
伴发小脑萎缩的疾病有很多种,其中最主要的是常染色体隐性遗传性疾病,也包括X连锁、染色体显性遗传疾病及线粒体疾病。随着医学的发展,检查技术的完善,对这些疾病的认知也在不断增加,尤其是在基因学领域更是突飞猛进。但是,要为这类患者制定合适的临床、基因检查仍是一个挑战,要想准确诊断这些疾病仍然非常困难。
神经影像学是一个非常重要的初期检查方法,尤其是颅脑MRI可以提供很多重要的信息,包括小脑及小脑以外结构的异常。
国外有一些关于小脑萎缩的影像学表现方面研究,国内仅有少数单一疾病的相关研究,横向归纳性的研究还是空白。我们的目的是依据MRI征象对不同伴发小脑萎缩的疾病进行分类,指导临床进一步检查,从而缩短疾病的诊断流程。
1 资料与方法
1.1 临床资料
我们回顾性分析了自1998—2012年经MRI确认为小脑萎缩的患儿,定义为小脑脑沟增宽、四脑室扩大、小脑体积缩小。年龄为新生儿至14岁。
排除标准:①后颅窝的肿瘤、手术后、出血、缺血被排除在外;②非对称性萎缩被认为是继发损伤被排除;③后颅窝畸形也被排除,例如Dandy-Walker、Arnold-Chiari畸形、Jubert综合征等。
最后共111例患者列入了我们的研究范围,其中男62例,女49例,76例病因明确,35例原因不明。除影像资料外,我们详细收集相关的临床资料,包括性别、发病年龄、确诊年龄、临床表现、家族史;实验室检查,包括代谢检查、免疫学检查、细胞、分子遗传学检查、肌电图、脑电图、皮肤或肌肉活检。
1.2 检查方法
每位患者至少进行1次MRI(1.5T或3.0T)检查,扫描序列包括:①矢状位T1WI,②横轴位T1WI、T2WI和/或T2-Flair,③部分患者进行了冠状位T2WI和/或T2-Flair扫描以及CT检查。
1.3 图像分析
所有结果由2位临床工作10年以上神经影像学医师对MRI表现进行评价。主要包括:①小脑萎缩的部位、程度;②小脑皮层T2WI信号异常;③小脑白质或齿状核的异常;④脑干萎缩及信号变化;⑤幕上白质、皮质及基底节的异常。
1.4 统计分析
应用SPSS 19.0对数据进行统计学分析,我们基于不同MRI征象使用最远距离、完全连接法对不同疾病进行系统聚类分析,最后确定最佳类集数为6类。
2 结果
2.1 不同疾病的MRI表现
111例患儿中经过实验室检查或临床活检共确诊76例,占总体68.4%。
线粒体疾病最多见,共23例,男15例,女8例。①Leigh’s病 7例(男 6例,女 1例)。MRI主要表现:4例重度萎缩,3例轻度萎缩。5例基底节受累(图1),1例丘脑受累;1例小脑皮层T2WI高信号;2例脑干受累;2例伴有大脑萎缩;3例伴有幕上白质T2WI高信号,其中1例内囊后肢受累。②Melas病5例(男4例,女1例),1例小脑蚓部轻度萎缩,有1例患者1年后复查显示小脑萎缩加重。3例患儿伴有大脑萎缩。2例基底节对称性钙化;4例患儿幕上局部皮层肿胀,分布无规律,呈长T1、长T2信号,DWI呈明显高信号。③呼吸链缺陷11例(男5例、女6例),MRI表现:6例轻度小脑萎缩,5例重度萎缩;1例小脑皮层T2WI高信号;3例脑干受累;基底节受累4例;幕上白质营养不良5例;大脑萎缩3例。
幼儿神经轴索营养不良(Infantile neuroaxonal dystrophy,INAD)共 13例,男 8 例,女 5例,均确认为PLA2G6基因突变。MRI主要表现:8例为轻度萎缩,5例为重度萎缩;2例伴有脑干萎缩;3例伴有大脑萎缩,1例伴有胼胝体萎缩;2例小脑半球皮层出现T2-Flair高信号(图2);7例伴有幕上脑白质T2WI信号增高;1例伴有双侧苍白球铁沉积。
神经元蜡样脂褐质沉积症(Neuronal ceroid lipofuscinosis,NCL)共 10 例,男 3 例,女 7 例(2 例是双胞胎姐妹)。其中婴儿型2例、晚婴型4例、少年型4例。MRI主要表现:重度小脑萎缩6例,轻度小脑萎缩4例;5例伴有大脑萎缩;9例幕上白质T2信号增高;3例基底节T2WI高信号;4例丘脑T2WI低信号(图 3)。
白质营养不良(Cockayne)综合征共4例,均为女性。MRI主要表现:均呈弥漫小脑重度萎缩;1例基底节T2WI高信号;1例小脑齿状核钙化,3例基底节钙化(图4);4例均表现幕上白质T2WI信号增高,集中在侧脑室周围深部白质;2例伴大脑萎缩。
肝豆状核变性5例,男4例,女1例。MRI主要表现:重度萎缩3例,轻度萎缩2例,小脑半球萎缩较蚓部明显;5例均可见基底节、丘脑对称T2WI高信号(图5);3例幕上白质局部T2WI信号增高;1例大脑萎缩。
异染性脑白质营养不良(Metachromatic leukodystrophy,MLD) 3例,男 1例,女 2例。MRI主要表现:小脑轻度萎缩2例,重度萎缩1例;均出现深部脑白质长T1、长T2信号影,从侧脑室表面到外周条纹状交错排列,即“豹纹征”,DWI示病变区水分子运动下降(图6)。
毛细血管扩张症性共济失调3例。MRI主要表现:均为弥漫重度萎缩,1例随访结果显示小脑萎缩加重,1例侧脑室前后角旁长T1长T2信号。
肾上腺脑白质营养不良(Adrenoleukodystrophy,ALD)3例,均为男孩。MRI主要表现:1例轻度萎缩,以上蚓部为著,2例弥漫重度萎缩;1例小脑齿状核T2WI对称信号增高;3例脑干皮质脊髓束均受累;2例丘脑T2WI高信号;均可见幕上白质大片长T1、长T2信号影;1例伴有大脑萎缩。
佩-梅氏病(Pelizaeus-Merzbacher disease,PMD)2例,佩-梅样病(Pelizaeus-Merzbacher-like disease,PMLD)1例。 MRI上两者表现非常相似,主要表现为弥漫白质长T1长T2信号,累及内囊、胼胝体及桥脑;PMLD患者伴大脑萎缩。
黏多糖贮积病Ⅰ型1例,男孩,4岁。MRI主要表现为小脑半球轻度萎缩,幕上深部白质区血管间隙增宽,大脑轻度萎缩。
甲基丙二酸血症1例,女孩,3岁。MRI主要表现为全小脑重度萎缩,双侧苍白球对称性长T1、长T2信号,脑室旁白质T2信号增高,大脑重度萎缩。
溶酶体转译后变体缺陷1例,男孩,4岁。MRI主要表现为全小脑重度萎缩,内囊后肢、幕上中央白质T2信号增高。
遗传性橄榄体桥脑小脑萎缩(Olivopontocerebellar atrophy,OPCA)、脊髓小脑萎缩症(Spinocerebellar ataxia,SCA)各 1 例,均为男孩。MRI表现为小脑半球轻度萎缩,SCA患者脊髓纤细。
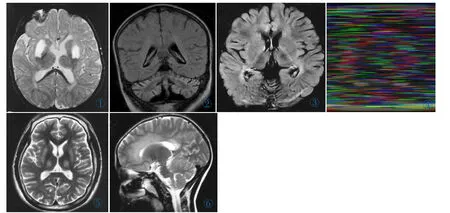
图1 女,1.5岁,Leigh’s病,双侧壳核对称长T2信号,侧脑室前后角旁白质长T2信号。 图2 男,6岁,INAD,小脑蚓部重度萎缩,小脑皮层T2-FLAIR高信号。 图3 女,4岁,NCL,小脑重度萎缩,侧脑室周围白质、内囊后肢T2WI高信号,双侧丘脑对称T2WI低信号。 图4女,4岁,Cockayne综合征,双侧基底节对称性钙化。 图5 男,12岁,肝豆状核变性,双侧壳核、丘脑对称性T2WI信号增高。 图6男,9岁,MLD,小脑轻度萎缩,脑白质弥漫脱髓鞘,“豹纹”征,病变累及胼胝体。Figure 1. A 1.5-year-old female with Leigh’s disease.High T2signal intensity is shown in bilateral putamen and white matter around the ventricle. Figure 2. A 6-year-old male with INAD.Severe atrophy occurs in the cerebellar vermis,with high T2-FLAIR signal in the cerebellar cortex. Figure 3. A 4-year-old female with NCL.Severe atrophy occurs in the cerebellum,with high signal in white matter around the ventricle and internal capsule,and low signal in bilateral thalamus. Figure 4. A 4-year-old female with Cockayne syndrome.Symmetrical calcification is seen in the bilateral basal ganglia. Figure 5. A 12-year-old male with Wilson disease.High signal intensity is seen in the bilateral putamen and thalamus. Figure 6. A 9-year-old male with MLD.Mild cerebellar atrophy is seen with diffuse demyelination in the white matter.The “leopard” sigh can be seen and the corpus callosum is involved.
未确诊病例共35例,占总体31.5%,男19例,女16例。MRI表现小脑轻度萎缩9例,重度萎缩26例;弥漫小脑萎缩24例,单纯小脑蚓部萎缩11例。
2.2 依据影像表现分组
我们把所有的影像表现分了7个组,以利于进一步鉴别。①单纯小脑萎缩:共33例,未确诊病例26例,INAD 1例,线粒体疾病2例,共济失调毛细血管扩张症2例,OPCA 1例,SCA 1例。②小脑萎缩伴皮层T2WI或FLAIR高信号:共6例,未确诊病例1例,INAD 4例,线粒体病1例。③小脑萎缩伴小脑白质或齿状核异常:共11例,未确诊病例2例,线粒体疾病 3例,INAD、Menkes氏稔毛综合征、MLD、ALD、PMD、纹状体小脑钙化和Cockayne综合征各1例。④小脑萎缩伴脑干异常:共18例,未确诊病例4例,线粒体疾病5例,ALD 3例,PMD 2例,PMLD、MLD、甲基丙二酸血症伴同型半胱氨酸尿症、肝豆状核变性各1例。⑤小脑萎缩伴幕上白质异常:共52例,未确诊病例14例,NCL 9例,线粒体疾病8例,INAD 9例,ALD 3例,MLD 3例,肝豆状核变性2例,Menkes氏稔毛综合征2例,溶酶体转译后变体缺陷、黏多糖增多症各1例。⑥小脑萎缩伴幕上白质髓鞘化不良:共14例,未确诊病例4例,Cockayne综合征3例,Menkes氏稔毛综合征2例,甲基丙二酸血症1例,PMD 2例,PMLD 1例。⑦小脑萎缩伴基底节或丘脑异常:共35例。基底节T2WI信号增高14例,未确诊病例2例,NCL 3例,线粒体疾病9例,肝豆状核变性5例,脑炎2例,甲基丙二酸血症、ALD、Menkes氏稔毛综合征各1例;丘脑T2WI信号增高16例(8例不伴基底节异常),未确诊2例,NCL 2例,线粒体疾病5例,ALD 2例,肝豆状核变性5例;2例NCL丘脑T2WI低信号;1例INAD苍白球T2WI低信号;基底节钙化6例。
2.3 聚类分析
①类集1,MRI表现为神经系统广泛受累,以深部灰质为主,主要包括线粒体疾病。②类集2,MRI表现为小脑重度萎缩,伴幕上白质脱髓鞘,主要集中在侧脑室周围白质、视放射及内囊后肢,主要包括INAD(8 例)和 NCL(8 例)。 ③类集 3,MRI表现为双侧基底节及丘脑对称性T2WI高信号,主要包括肝豆状核变性(5例)和线粒体疾病(3例)。④类集4,MRI表现为基底节对称性钙化,伴幕上白质髓鞘化不良,主要包括Cockayne综合征。⑤类集5,MRI表现为幕上白质弥漫脱髓鞘,同时伴有脑干及小脑白质T2WI信号增高,主要包括ALD、MLD、Menkes氏稔毛综合征。⑥其他疾病被分入类集6,样本量较少,MRI征象无明显特征性。
3 讨论
我们回顾分析了5年内到北京大学第一医院就诊的伴有小脑萎缩的患儿,报告了相关疾病的发生率。虽然检查技术发展非常迅速,尤其是基因领域,但还是有相当多的患儿(30.3%)没有明确诊断。在确诊病例中,线粒体疾病最多见(27.7%),与国外的一些研究报道[1-4]相似,其后是INAD、NCL、肝豆状核变性、Cockayne综合征、毛细血管扩张性共济失调等。
单纯小脑萎缩中,不明原因的比例最高,说明没有特异征象的小脑萎缩很难诊断。而在已知的病因中常染色体隐性遗传小脑共济失调最多见,主要包括Friedreich共济失调和毛细血管扩张性共济失调[5-8]。Friedreich共济失调临床特点为进行性共济失调、心肌病、下肢深感觉丧失、腱反射消失以及锥体束征,常伴骨骼畸形。主要病理基础是神经变性。诊断主要依据临床表现,尤其心脏受累较多见。生化检查常提示血中胆红素增多,胰岛素下降。毛细血管扩张性共济失调基因突变位于染色体的11q22~23,即负责编码蛋白激酶的ATM基因突变导致发病。发病多在2~4岁,病理特点为小脑萎缩,主要累及小脑蚓部,半球受累相对少见,浦肯野细胞、颗粒细胞及蓝状细胞减少。诊断主要依靠临床表现,即小脑共济失调、毛细血管扩张症及反复的肺部感染。实验室检查主要有外周血中淋巴细胞百分比降低,免疫球蛋白IgA、IgE缺乏或降低,血清甲胎蛋白增高。也包括少量早发型遗传性小脑共济失调,如IOSCA等,本次研究中有1例,1~2岁发病,基因突变位于染色体10q22.3~q24.1,主要的病理基础为神经元丢失,确诊可行PCR分析,用外周血白细胞检测相应基因CAG扩增,证明SCA的基因缺陷[9-11]。
小脑萎缩伴小脑皮层高信号中,INAD的比例最高(66.6%),而且发生率也较高(30.7%),因此当出现小脑萎缩伴小脑皮层高信号时,要首先考虑该病。INAD是由于PLA2G6基因突变导致的,神经病理显示神经纤维特异性球样扩张[12-14]。临床表现主要为快速进行性张力减低,对称性锥体束征,腱反射亢进,痉挛性四肢瘫痪,运动及认知能力的明显倒退,视力受损,眼球震颤。病理基础主要是神经元丢失、皮层萎缩、脱髓鞘、胶质增生,神经电生理检查提示神经源性损伤。其他疾病还有线粒体疾病、Marinesco-Sjögren综合征、先天性糖基化障碍、溶酶体疾病等,但出现率较低。线粒体疾病需要进行光谱酶学检查及线粒体功能分析。Marinesco-Sjögren综合征的诊断需要依靠家族史、临床表现及影像学表现[15-16],没有较特异的诊断性检查,先天性糖基化障碍需要做血浆转铁蛋白等电聚焦测定,转氨酶及一些溶酶体酶升高。
小脑萎缩伴发脑干异常并不多见,即使是在病程的后期。伴发脑干萎缩的有:黏多糖贮积症、Salla病、脊髓小脑共济失调及消散性脑白质病。脑干信号异常多见于线粒体疾病,也可见于肝豆状核变性、ALD等,多为灰质核团。如果怀疑线粒体疾病,可以进行血生化检查,如血乳酸、丙酮酸最小运动试验,线粒体呼吸链复合酶活性检测;活检可见肌细胞内线粒体堆积;基因检查主要注意容易突变的位点。肝豆状核变性诊断依据典型锥体外系症状、肝病体征、角膜K-F环和阳性家族史等诊断不难,实验室检查主要包括铜代谢相关检查、肝肾功能检查[17-21]。肾上腺脑白质营养不良的MRI表现较特异,即对称性后部脑白质长T2信号,边缘环状强化;另外也可进行内分泌功能检测及血浆、皮肤成纤维细胞VLCFA测定[22-25]。Salla病可以通过检测游离唾液酸水平以及SLC17A5基因来诊断[26]。
小脑萎缩伴发基底节及丘脑异常很常见,多表现为T2WI信号增高,例如丙酮酸激酶缺乏、Cockayne综合征、肝豆状核变性、L-2-羟基戊二酸尿症、NCL、INAD以及线粒体疾病。NCL可见丘脑T2WI信号降低,INAD苍白球T2WI信号降低(铁沉积所致),即“虎眼”征。Cockayne综合征及慢性进行性眼外肌麻痹(Keams-Sayre syndrome,KSS)、线粒体肌病脑病伴乳酸中毒及中风样发作(Mitochondrial encephalomyopathy,lactic acidosis,and stroke-like episodes,MELAS)可以出现基底节钙化,肝豆状核变性的“熊猫眼”征较特异。NCL的诊断主要依靠临床表现、病理检查结果和基因检查,其中病理检查发现病理性脂褐素颗粒是诊断的金标准[27-30]。Cockayne综合征的诊断主要依靠临床表现,如生长障碍、进行性神经功能异常等,确诊方法为基因诊断,尤其是CSA和CSB基因缺陷[31-33]。
大多伴有小脑萎缩的先天性疾病都会伴有幕上白质的异常、髓鞘化不良或脱髓鞘。髓鞘化不良的疾病主要有:PMD、Salla病、丙酮酸激酶缺乏及糖基化障碍。其中PMD最明显,仅在内囊、视放射和放射冠近端出现T1WI高信号,PMD临床诊断主要依据典型的临床表现及头颅影像学检查,确诊依靠分子遗传学研究。临床上遇到男性患儿以眼球震颤起病,肌张力低下、共济失调和进行性运动功能障碍要考虑该病,进一步行PLP1基因检查以确诊[34-36]。丙酮酸激酶缺乏的诊断依赖于红细胞丙酮酸激酶活性的测定。脱髓鞘的疾病主要有:Cockayne综合征、NCL、肝豆状核变性、ALD、MLD及线粒体疾病,多表现为侧脑室周围的深部脑白质、侧脑室三角区以及视放射长T2信号。其中只有Cockayne综合征皮层下U型纤维早期即可受累。其中ALD为炎性脱髓鞘,增强扫描看见环状强化。MLD表现为垂直侧脑室表面正常与异常髓鞘化交叉排列,即“豹纹”征,相对特异。MLD的临床表现无特异,诊断需要依靠病理或基因检查。尿沉渣发现大量异染颗粒可初步诊断。检测血白细胞及皮肤成纤维细胞中ARSA活性可确诊该病[37]。
我们对本次研究所包含的疾病依据MRI征象进行了聚类分析。结果显示,虽然导致儿童小脑萎缩的疾病非常繁杂,但是还是可以对不同疾病进行分类的。
类集1只包含线粒体疾病,由于本病包含一组疾病,影像学表现复杂多样,神经系统受累广泛。个别综合征的影像学有一定特征,例如Leigh’s综合征主要累及深部灰质核团,尤其是壳核,有研究指出几乎100%受累;MELAS最显著的影像学特点是多发散在卒中样异常信号 ,累及皮层和皮层下白质,并不按解剖血管分布,病变位置不固定,呈游走性;KSS的特征性表现是基底节对称性钙化,皮质下U型纤维受累,而脑室旁白质多正常[38]。还有很多线粒体疾病无明显特征性MRI表现,因此我们认为任何有神经系统症状而无明确特异性MRI表现的患儿都不能排除线粒体疾病,尤其是有深部灰质异常的患儿都应该考虑该病。
类集2包括了NCL和INAD两种疾病。主要表现为小脑重度萎缩及幕上白质脱髓鞘。NCL小脑重度萎缩占80%,INAD小脑重度萎缩占62.5%,这可能与病程有关,因为两者的小脑萎缩均呈进行性。幕上白质脱髓鞘的发生率分别为80%和69.2%,主要位于侧脑室周围及视放射,内囊后肢也多受累,可见锥体束多受累。
类集3只包含肝豆状核变性,MRI主要特征是双侧基底节、丘脑均受累,呈对称性T2WI高信号,发生率为100%。病理基础可能是水肿、胶质增生、脱髓鞘、神经元坏死或囊变。其他疾病很少出现基底节、丘脑均受累,本次研究中只有ALD和NCL各1例。
类集4只包括Cockayne综合征,主要MRI特征为基底节对称性钙化,伴幕上白质髓鞘化不良。出现基底节对称性钙化的疾病并不多见,例如巨细胞病毒感染的钙化多位于脑室周围室管膜下,常伴脑回皮层异常;Aicardi-Goutie’eres综合征的钙化多较小,多位于基底节及侧脑室周围白质;其他髓鞘化异常的疾病(如PMD)很少出现钙化;线粒体疾病的表现可以与Cockayne综合征表现相近,但是重度的进行性的脑萎缩多见于后者。
类集5包括ALD、MLD、Menkes氏稔毛综合征,主要MRI征象为幕上白质脱髓鞘、脑干异常及小脑白质异常。反映了这3种疾病的皮质脊髓束受累非常多见,但需要指出的是,ALD幕上白质脱髓鞘为炎性脱髓鞘,并且累及部位较典型,即多位于后部白质,而MLD与Menkes氏稔毛综合征脱髓鞘范围较弥漫[39]。另外,Menkes氏稔毛综合征有一个较典型的征象,即硬膜下积液,本次研究中有3例,但是该征象并未列入本次研究范围内。
其他疾病包括在类集6中。由于相关疾病的样本量较小,可能并不具有很好的代表性,但是也可以看到一些特点,如PMD和PMLD主要表现弥漫的髓鞘化不良;毛细血管扩张性共济失调、SCA及OPCA仅表现小脑萎缩,不伴其他征象;黏多糖增多征表现幕上白质血管间隙增宽。可以为以后的研究提供一定的方向。
影像学与临床资料相结合,可以为儿童期伴发小脑萎缩疾病制定一个诊断路径;神经影像学模型在疾病鉴别诊断及指引进一步检查上起着至关重要的作用。本研究的不足之处:①在于回顾分析及潜在的偏倚性,未确诊病例较多。②本次研究涵盖的病种并不全面,某些疾病的样本量过少,还需要我们继续收集总结。③每种疾病的MRI表现并不是绝对的,在不同个体及不同病程表现会有差异。④小脑萎缩与小脑发育不良在某种疾病中很难区分。
猜你喜欢
中国医药导报(2022年28期)2022-11-25
保健与生活(2022年9期)2022-05-06
中国医学影像技术(2022年3期)2022-03-18
中国典型病例大全(2022年1期)2022-01-10
健康博览(2021年7期)2021-08-16
老年医学研究(2021年6期)2021-03-09
中国卫生标准管理(2020年21期)2020-11-27
保健与生活(2020年11期)2020-06-23
中国医学影像技术(2020年11期)2020-01-13
创新作文(小学版)(2019年4期)2019-07-24
