
Review on Li-insertion/extraction Mechanisms of LiFePO4 Cathode Materials①
2019-01-05 08:07WUYiFngCHONGShoKunLIUYongNingGUOShengWuBAILiFengLIChengShn
结构化学 2018年12期
WU Yi-Fng CHONG Sho-Kun LIU Yong-Ning GUO Sheng-Wu BAI Li-Feng LI Cheng-Shn
Review on Li-insertion/extraction Mechanisms of LiFePO4Cathode Materials①
WU Yi-FangaCHONG Shao-KunbLIU Yong-Ningb②GUO Sheng-WubBAI Li-FengaLI Cheng-Shana
a(710016)b(710049)
The work distills the main mechanisms during the lithium insertion/extraction of LiFePO4cathode materials. The “diffusion-controlled” and “phase-boundary controlled” mechanism are especially illustrated. Meanwhile, some recent observation and analyses byoron the Li-insertion/extraction of LiFePO4are summarized and prospected.
Li-ion battery, LiFePO4, Li-insertion/extraction, mechanism, review;
1 INTRODUCTION
Lithium iron phosphate (LiFePO4) has become one of the most promising Li-ion battery cathode materials due to its high safety performance, long cycle life, good platform performance, low cost and environmental friendly[1-11], which is of great signifi- cance for the development of electric vehicle industry[12-13].
Although considerable progress has been made in the development and production of the power battery of LiFePO4, and it has gradually entered the practical orbit, its price is still high, and the high rate charge/discharge capability still needs to be further improved[14-32]. For example, when LiFePO4battery is used in electric vehicles, its power cannot be compared with that of traditional fuel engines, which influences the journey, top speed, acceleration and climbing capabilities of electric vehicles.
In order to promote the development of LiFePO4battery and improve its high rate charge/discharge capability, it is necessary to study the mechanism of Li-insertion/extraction of LiFePO4. The mechanism involves the complex process of physical chemistry and reaction kinetics, so in-depth study and unders- tanding of its basic behavior will greatly help to improve the rate capability of LiFePO4cathode materials.
2 MAINSTREAM VIEWS
In this work, we illustrate two main mechanisms including the “diffusion-controlled”and “phase- boundary controlled” mechanisms.
2. 1 Diffusion-controlled mechanism
In this view, the mechanism of Li-insertion/extrac- tion of LiFePO4is “diffusion controlled” model. The phase transition of lithium follows the Radial model or Mosaic model in the Li-insertion/extraction pro- cess[33], that is, the shell of one phase contains the core of another phase, and diffusion occurs throu- ghout the shell moving along the interface. Com- pared with the Radial model, the Mosaic model thinks the Li-insertion/extraction can occur anywhere within a LiFePO4particle. Fig. 1 (a) and (b) show the schematic representations of two possible models (Radial model or Mosaic model) for Li-inser- tion/extraction in a single particle of a LiFePO4during charging and discharging.
In the “diffusion-controlled” model, one of the most compelling evidences is the result of tempera- ture-controlled XRD spectrum, indicating that LiFePO4exhibits thermally activated solid-solution behavior in the composition range[34]. As shown in Fig. 2, the phases of Li1–yFePO4and LiFePO4at room temperature present the broadening at 210 ℃, and then become completely solid solution at 290 ℃.
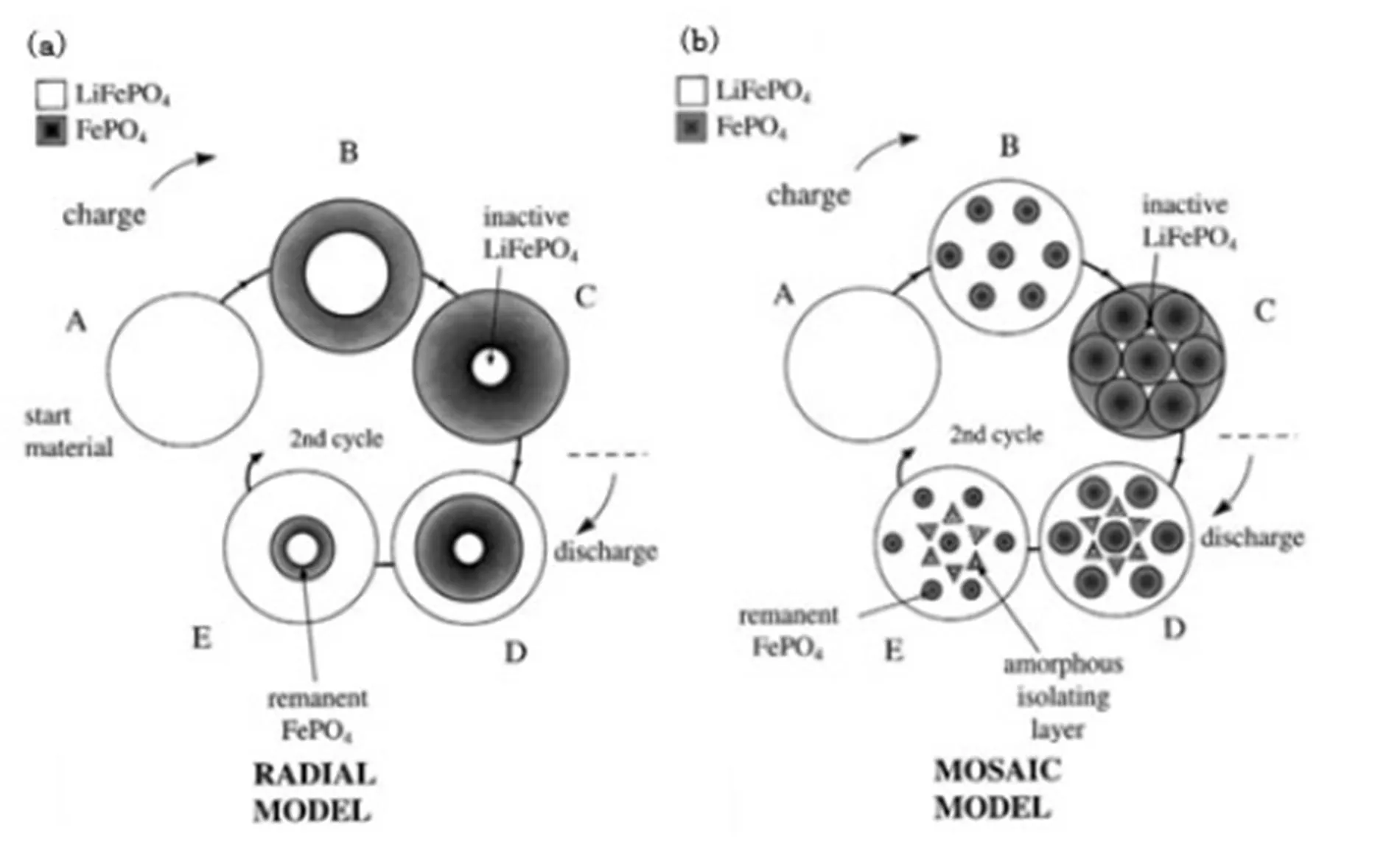
Fig. 1. Diffusion-controlled type (a) a “radial model” (b) a “mosaic model”
Fig. 2. Temperature-controlled XRD patterns (Co) of Li0.68FePO4under N2
Another strong evidence is the result of tempera- ture-controlled Mӧssbauer spectrum[35]. Some deli- thium samples were carried out, suggesting the solid solution marked with blue line increased, accompa- nied by the reduction of the mother source Fe2+and Fe3+at elevated temperature (Fig. 3). Both studies found solid solution phases appear in their samples, implying that the Li-insertion/extraction is controlled by the diffusion related to the concentration gradient.
Malik[36]proposed a single-phase dynamic model of the phase transition of LiFePO4using the first principle and Monte Carlo method (Fig. 4). They reported that LiFePO4might be embedded in a solid solution based on the free energy calculation of the equilibrium state, supporting the “diffusion-con- trolled” model. Furthermore, although the two-phase equilibrium characteristics of the LiFePO4and FePO4phases are obvious for some Li-insertion of LiFePO4, the phase transition kinetics of LiFePO4may exhibit an alternative single phase transition, which can occur at a very low overvoltage without nucleation and growth. Since it can occur at very low overvol- tage, the single-phase kinetic model explains good LiFePO4has excellent rate capability.

Fig. 3. Temperature-dependent Mӧssbauer spectra of Li0.55FePO4. The solid lines (green, Fe2+; red, Fe3+; blue, solid solution) are individual components that contribute to the overall fitting of the spectra. Transformation of each of the two-phase samples into LixFePO4single phases is initiated at 200 ºC, and completed at 350 ºC[38]
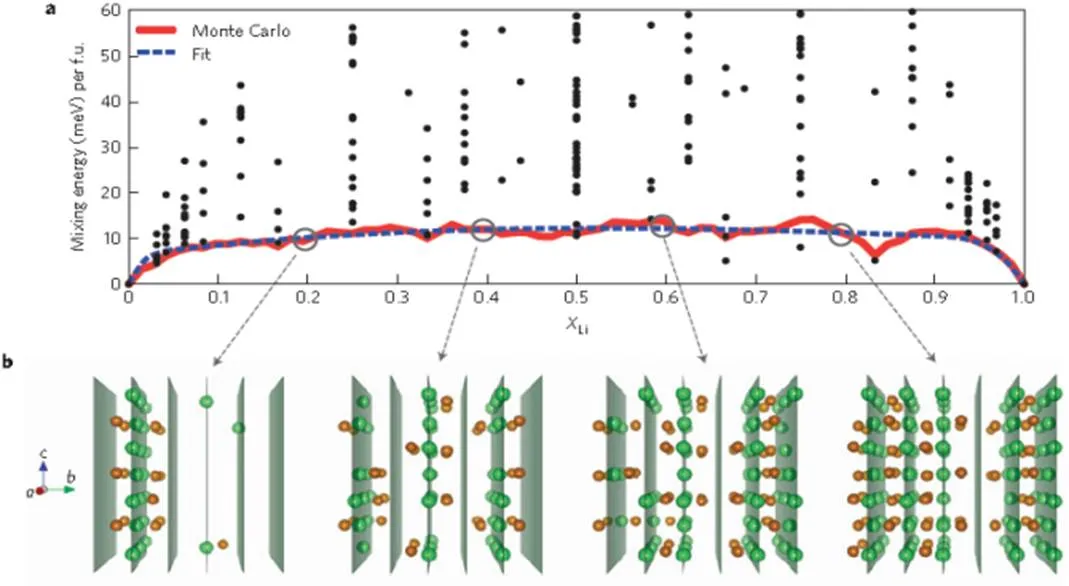
Fig. 4. Free energy and atomic configurations along the single-phase LiFePO4transformation path. (a) Zero-temperature mixing energies (black circles) calculated from the first principles of 245 different Li/vacancy and electron/hole configurations in LiFePO4(0≤≤1) show the existence of several low formation energy structures. (b) Snapshots of Li (green atoms) and Fe2+(brown atoms) configurations in Monte Carlo simulationsat room temperature forLi= 0:2, 0.4, 0.6 and 0.8 show the succession of single-phase states with some local ordering[39]
2. 2 Phase-boundary controlled mechanism
Although the above model can perfectly explain the charging/discharging of LiFePO4battery, it is questioned by another view of “phase-boundary controlled”. This is mainly because some researchers observed that FePO4domain was found in some LiFePO4particles in Li-insertion/extraction process, and the interface area was not the solid solution, but the superposition of LiFePO4and FePO4phases.
Based on these observations and analysis, some alternative phase-interface controlled mechanisms, such as the spinodal-decomposition mechanism[37]and the domino-cascade mechanism[38], were pro- posed, as marked in Figs. 5 and 6, respectively. In the amplitude modulation decomposition, the component at the peak is higher than the average one, whereas the component at the trough is lower than the average one. However, the crystal structures in the Li-rich and Li-poor regions were the same, except that the composition fluctuated slightly. Both mechanisms consider that the Li-ion insertion or extraction involves the coordinated movement of lithium ions throughout the phase interface, and the main difference between them is the velocity of the phase interface. The domino-cascade mechanism held the phase interface was moving rapidly, and the mixed phases of LiFePO4and FePO4were not detected in some delithiated particles. However, the velocity of phase interface is relatively low in the spinodal- decomposition mechanism, and multi-domains and phase interfaces can be observed in partially deli- thiated particles.
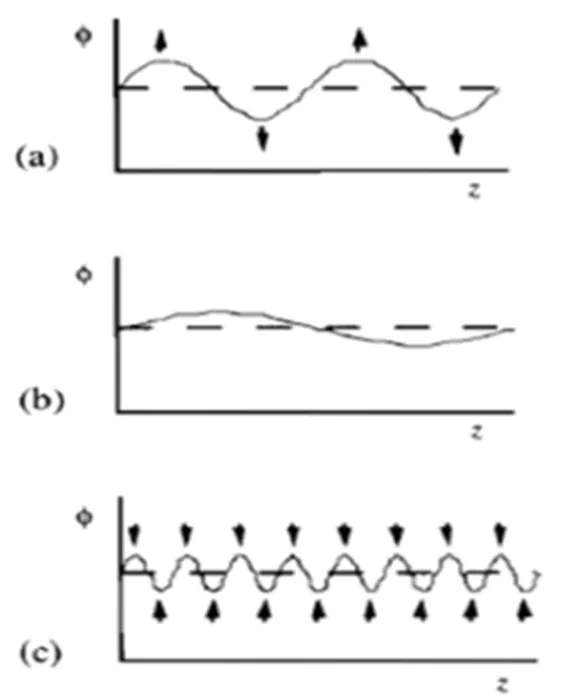
Fig. 5. Schematic view of the ‘Spinodal-decomposition’ mechanism. (a) An intermediate length scale grows the fastest and dominates the pattern of phase separation. (b) A long-wavelength fluctuation grows relatively slowly, as the distances that the material has to diffuse from troughs to peaks are relatively large. (c) Too much new interface is created, with a correspondingly large free energy penalty
Fig. 6. Schematic view of the ‘domino-cascade’ mechanism for the lithium deintercalation/intercalation mechanism in a LiFePO4crystallite. (a) Scheme showing a view of the strains occurring during lithium deintercalation. (b) Layered view of the lithium deintercalation/intercalation mechanism in a LiFePO4crystalline
One of the strongest evidences supporting the “phase-boundary controlled” mechanisms is the high-resolution electron energy loss spectroscopy (EELS, O-K edge) of the delithium sample Li0.45FePO4[39], suggesting the spectral line of the interface is a linear combination of LiFePO4and FePO4spectral lines. Hence, we can deduce that the interface is a two-phase coexistence region, rather than a solid solution, as presented in Fig. 7. It can also be seen from Fig. 7 that the interface width of LiFePO4and FePO4phases is 12 nm.
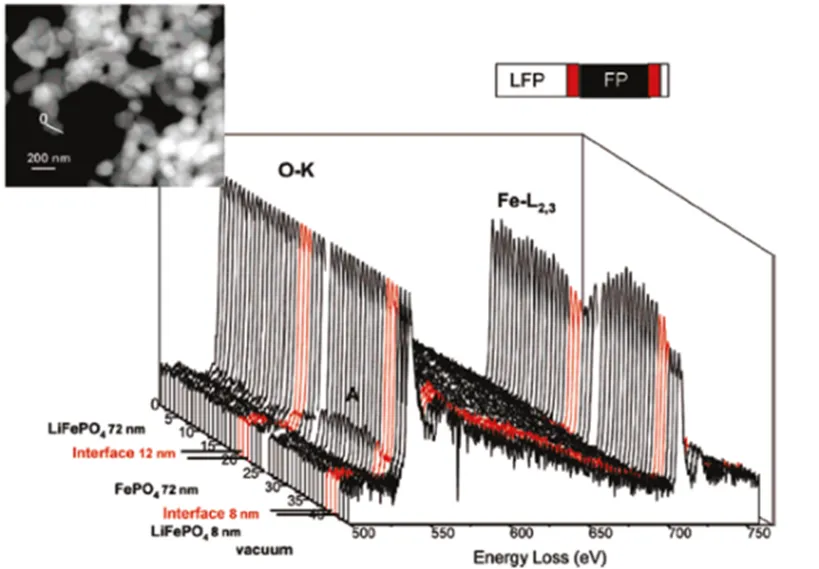
Fig. 7. STEM HAADF image of the chemically delithiated sample Li0.45FePO4with the analysis line and the 3D representation of EELS spectra recorded along that line. Note that the EELS spectra of the interface are represented in red
Another strong evidence is the high-resolution transmission electron microscopy (HRTEM) spec- trum[40], which shows multi-domains and phase interfaces can be observed in partially delithiated LiFePO4particles. As shown in Fig. 8, a small angular grain boundary along theaxis is formed because of the dislocation motion, indicating the dynamic process is caused by the dislocation and the movement of nucleation front, rather than the diffusion related to the concentration gradient.
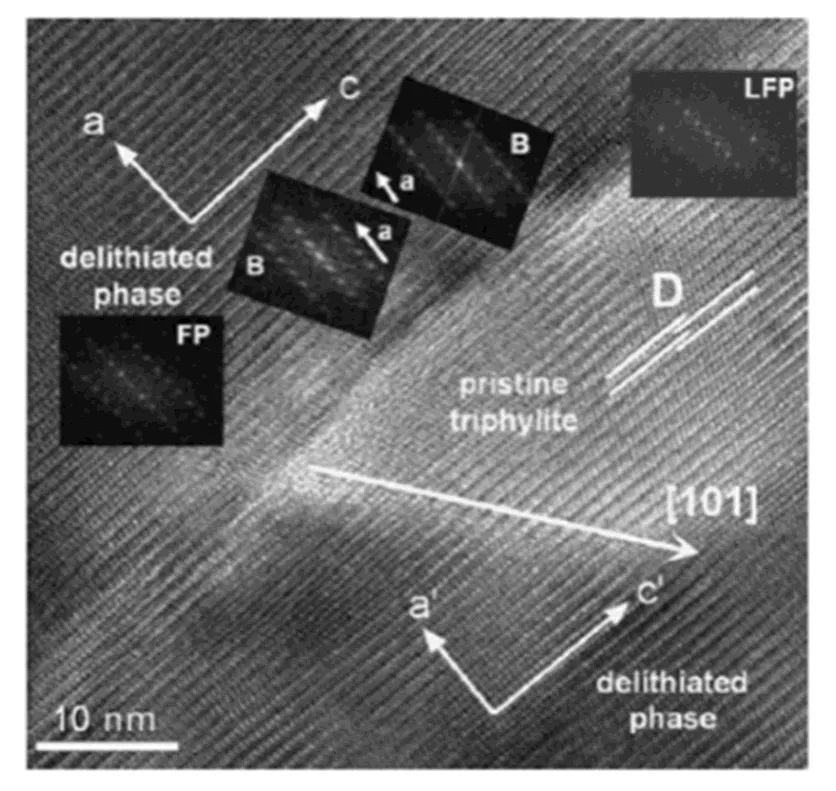
Fig. 8. HRTEM image of a partially delithiated LiFePO4particle showing a LiFePO4domain (LFP) in theplane surrounded by FePO4phase (FP) with narrow interface layers consisting of two crystal phases
Allen[41]further proved the viewpoint of “phase- boundary controlled” with the Avrami-Johnson- Mehl-Eroofev equation to analyze the delithiated dynamics.
= 1 – exp(–)(1)
Whereis the volume fraction of LiFePO4,is the rate constant parameter, andis an exponent whose value is dependent upon the geometry of the transformation. The Avrami exponentincreases with the dimensionality of growth. Typically, anvalue of 1~2 is indicative of one-dimensional growth. Allen obtained the Avrami exponent value of 1, elucidating the phase transition of Li-inser- tion/extraction follows the one-dimensional growth mechanism and belongs to the “phase-boundary controlled” type.
The XPS spectra[42]also showed that, upon char- ging, the ratio of Fe3+/Fe2+on the surface of LiFePO4continued to increase, rather than presenting an unusually large rate of Fe3+/Fe2+, which conforms to the rule of “phase-boundary controlled” model. This can be explained by the fact that most Fe3+ionsshould be gathered on the surface of LiFePO4according to the core-shell model. A large rate of Fe3+/Fe2+should appear in the XPS analysis, but as noted above, the results are not expected. Fig. 9 shows the schematic diagram of XPS spectra according to different models.

Fig. 9. Scheme to describe the expected XPS spectra according to different models[44]
Recently, Bai[43]also proposed a surface-limited Li-insertion model for anisotropic nanoparticles in non-equilibrium state with the phase field, and pre- dicted that phase separation could be inhibited at a certain critical current. Due to the reaction limitation of nanoparticles, if the applied and exchange currents are at the same order of magnitude, the surface overpotential of the nanoparticles can easily exceed the voltage limitation of the solid solution to prevent the thermodynamic driving force of phase separation. Phase separation will play a major role only if the current is low in very large particles. The amplitude- modulated decomposition or nucleation causes the phase boundary to move at a small current. When the current density is higher than a certain critical one, the amplitude modulation decomposition disappears and the particles are evenly filled, which can explain the high multiplier performance and long cycle life of nanometer LiFePO4. The theory holds that in the non-equilibrium state, the “diffusion-controlled” model plays a leading role in the nanoparticles, whereas the “phase-boundary controlled” model plays a dominate role in micron particles scale with a small rate discharge.
The above viewpoint was further confirmed by Oyama[44]using the potential step method and the calculation of Avrami exponent in the Kolmogorov- Johnson-Mehl-Avrami (KJMA) model. The results indicated that Li-insertion/extraction depends on the magnitude of the potential step and particle size, as shown in Fig. 10. When the particle size (< 100 nm) is small while the potential step (< 150) mV is large, the chronoamperometryresult shows the current monotonically declines, which is consistent with the KJMA model, suggesting that the Li-insertion/extrac- tion pathway is a non-equilibrium solid solution way. When the particle size of 203 nm is large while the potential step of 10 mV is small, the chronoam- perometry data show that the current increases at the moment and then gradually decreases, implying the Li-insertion/extraction is dominated by the electrode dynamics with the growth of new phase nuclei. Meanwhile, the chronoamperometry diagram is fitted with the KJMA model, and the Avrami exponent= 1conforms to the one-dimensional phase boundary model. Therefore, in this mechanism, in the non- equilibrium state, the Li-insertion/extraction mode of spherical particles of < 100 nm is the “diffusion- controlled” model dominated by solid solution, whereas the ones of > 100 nm are fitted with the “phase-boundary controlled” model.
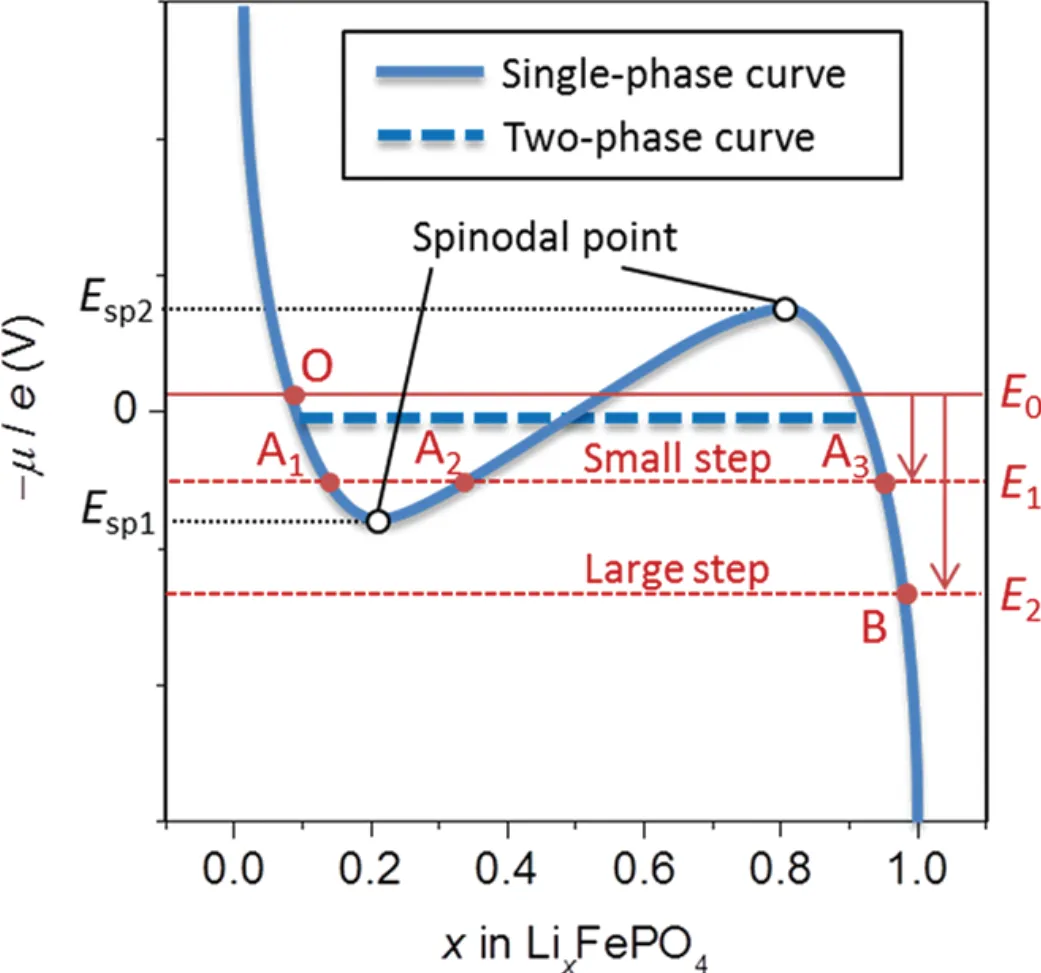
Fig. 10. Chemical potential profiles of LiFePO4, assuming a single phase reaction (blue solid line) and a two-phase reaction (blue dashed line). Symbols O, A1, A2, A3, and B denote the equilibrium states at different potentials (E0, E1, and E2). The white circles indicate spinodal points. In a potential region between two spinodal points (Esp1 and Esp2), there exists a metastable solid solution (A1) as well as a stable one (A3)[47]
3 RECENT ADVANCES
In recent years, the researches on the mechanism of Li-insertion/extraction mechanism of LiFePO4have never been stopped, especially with the deve- lopment ofobservation technique, the mecha- nism has been further investigated.
Gu[45]observed in situ for the first time that lithium was intercalated during the extraction process on the atomic scale with the aberration-corrected annular-bright-field (ABF) STEM (Fig. 11). The results show that the rest of Li ions occupies per- fectly the interlayer atoms along theaxis in the partial delithium nanowire (electrochemical deli- thium,= 65nm) LiFePO4, which is more inclined to the previously proposed “diffusion-controlled” mechanism.
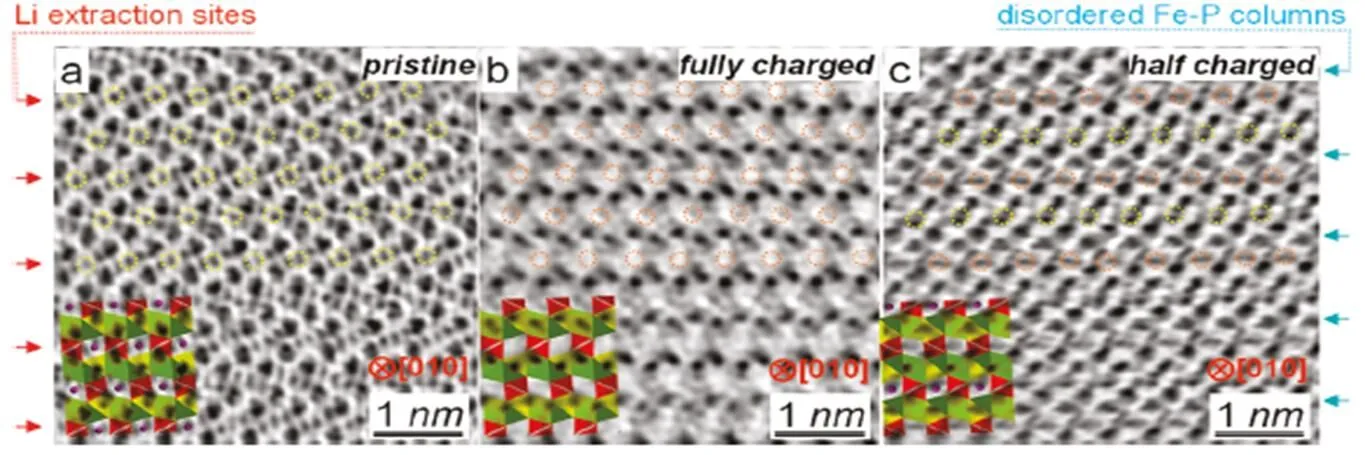
Fig. 11. Aberration-corrected annular-bright-field (ABF) STEM micrographs showing Li ions of partially delithiated LiFePO4at every other row (a) Pristine material with the atomic structure of LiFePO4shown as inset; (b) Fully charged state with the atomic structure of FePO4shown for comparison; and (c) Half charged state showing the Li staging[48]
Chang[46]found that the phase transition from FePO4to LiFePO4(or from LiFePO4to FePO4) was hysteretic during the extraction process of lithium of LiFePO4. Specially, this hysteretic behavior was also present at low medium magnification (C/10~1C) withSynchronous XRD spectrum. The inset in Fig. 12 shows the XRD peak intensities of LiFePO4(020) (■) and FePO4(020) (□) versus the measurement number in the process of discharging- rest-charging-rest (D-R-C-R). It can be seen that the FePO4phase is converted to LiFePO4phase until it is near the end of discharge. Similarly, LiFePO4phase is converted to FePO4until near the end of charge. The results indicate that, even in the metastable crystal structure (the charge/discharge is under non-equilibrium conditions), the characteristics of two phases of Li-insertion/extraction are obvious, which is more in line with the “phase-boundary controlled” model.

Fig. 12.synchrotron XRD patterns for charge/discharge test of the cell with an intermittent rest period. The subscripts F and L indicate, respectively, the reflections from the FePO4-and LiFePO4-structures. The bold curves mark the onset of different stages (C for charge; D, discharge; R, rest) within a test cycle. The inset shows the XRD peak intensities of LiFePO4(020) (■) and FePO4(020) (□) versus the measurement number[49]
Wang[47]studied the LiFePO4samples with 0.6- micron usingX-ray diffraction (XRD),X-ray absorption spectrometry (XAS) and pre- positional soft X-ray absorption spectrometry. As shown in Fig. 13, the contents of LiFePO4and FePO4phases deviated significantly from the linear region shown in the dotted line during the charging process through the Rietveldrefinement. This indicates that the phase transition lags behind the voltage curve in the partially delithiated Li1–FePO4. Studies on the combination ofand prepositional methods show that the phase transition is related to not only the composition of Li, but also the different nuclea- tion and growth mechanism of the new FePO4phase during charging, which supports the “phase-boun- dary controlled” model.

Fig. 13. Contents of LiFePO4and FePO4calculated by Rietveld refinement.in Li1-FePO4[50]
Wang[48]investigated the Li-insertion/extraction mechanism of multi- and single-particle systems byTXM-XANES spectrum. As shown in Fig. 14, for multi-particle nanometer FePO4aggre- gation, the transition from LiFePO4to FePO4phase is uniform at the slow delithium rate of 0.1 C, and more LiFePO4and FePO4exist in the form of mixed phase. These conclusions support the solid solution viewpoint where phase separation is inhibited in the Li-insertion/extraction process, which agrees with the “diffusion-controlled” mechanism. However, more LiFePO4and FePO4phases co-existed in the transformation process at the high delithium rate of 5 C, supporting the “phase-boundary controlled” mechanism. For the single-particle system, as shown in Fig. 15, the two phases of FePO4and LiFePO4co-existed in the micron scale particles revealed byfull-field TXM, and their mixed phases were not detected at the delithium rates of 0.02 and 1 C, respectively, supporting “domino-cascade” model of the “phase-boundary controlled” mechanism. In addition, the results also show the Li-inser- tion/extraction mechanism of LiFePO4is closely related to not only the particle size, but also the test conditions of sample such as discharge rate.
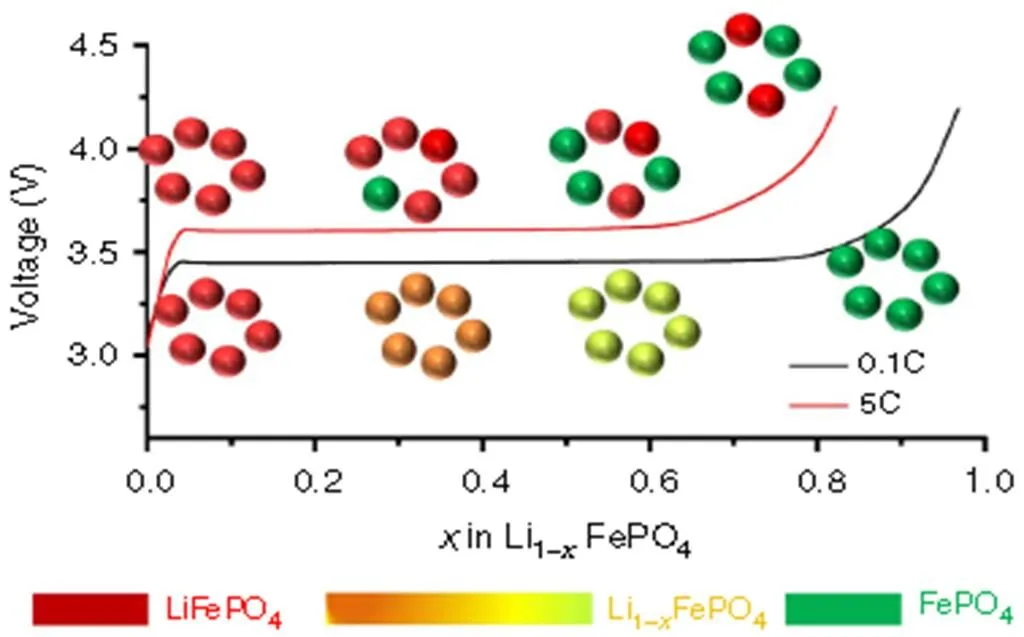
Fig. 14. Schematic illustration for multi-particle LiFePO4system. Phase transformation mechanism and intercalation pathway at fast and slow charging rates for multi-particle LiFePO4system. Each circle represents a multi-particle nano-LFP aggregation, not single particle[51]
Fig. 15. Schematic illustration for single-particle LiFePO4system. The models a and b and the corresponding actual particles represent the two typical delithiation processes revealed byfull-field transmission X-ray microscopy (TXM)[51]
Sharma[49]concluded the coexistence of solid solu- tion and two-phase transition behavior in the non- equilibrium samples of deep discharge usingneutron powder diffraction (NPD). In Fig. 16, the Rietveld refinement parameters of LiFePO4phase are represented by green cross, while those of FePO4phase are represented by black cross. The vertical black line is the starting point of solid solution reac- tion, while the vertical purple line is the transition curve from Li1–FePO to LiFePO4phases in chronological order. The shadow region is a two- phase region. This study shows the Li-inser- tion/extraction mechanism of LiFePO4is closely related to the state of the sample, which may be different when the sample is in equilibrium state and non-equilibrium state.
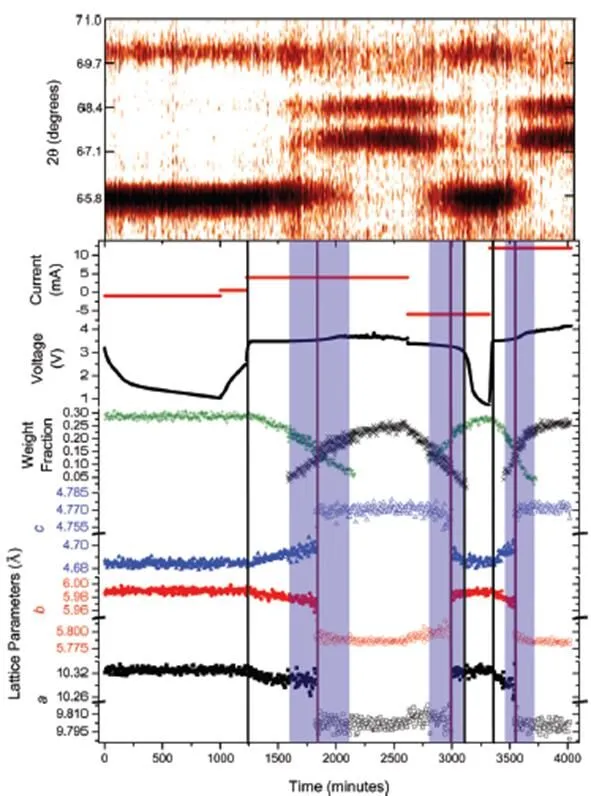
Fig. 16.neutron powder diffraction (NPD) data of the Li||LiFePO4battery and Rietveld-derived lattice parameters of the cathode[52]
Yu[50]investigated the Li-insertion/extraction mechanism of LiFePO4for the ellipsoid samples (300 nm) under the chemical and electrochemical delithium at different time (0.1~60 C) withXAS. Meanwhile, they obtained the value 1 of the Avrami exponent with the Avrami-Johnson-Mehl- Eroofev equation, which conforms to the one-dimen- sional “phase-boundary controlled” mechanism.
Niu[51]studied the dynamics of LiFePO4electrode of nanowire (200~400 nm) at the discharge rate of 1 C in the delithium process byHRTEM. They observed in situ the sub-lattice disordered solid solution area (SSZ) of lithium, which was completely different from the sharp LFP|FP boundary observed under other conditions. The solid solution region with a scale about 20 nm is so stable that it can exist for hundreds of seconds at room temperature. Since there is no dislocation in the solid solution area, greater cycle life and higher rate capability can be expected, which implies that the disordered solid solution area may dominate phase transition behavior in the non-equilibrium state (when higher current or voltage is used). The results indicate the sample of this scale obeys the “diffusion controlled” mecha- nism under non-equilibrium conditions.
Lim[52]observed the kinetic map of Li compo- sition distribution and insertion rate of the flaky LiFePO4particles (1 μm in wideness and 150 nm in thickness) using anscanning transmission X-ray microscopy(STXM). As shown in Fig. 17, the phase separation was inhibited as the lithium rate increase, and the insertion pathways of lithium converted from “phase-boundary controlled” (under 0.2 C rate) to “non-uniform domain area” and then to “uniform solid solution” (under 2 C rate). When the lithium rate increased to 2 C, the Li component distribution observedindicated Li is uniformly inserted into the particle, namely, the composition difference between each particle is very small, which is considered to be a solid solution behavior. The conclusion is in line with the “diffusion controlled” mechanism. The depen- dence of composition distribution on the ratio makes it easier to form uniform solid solution phase in the process of lithium formation, while the process of delithium is less uniform. The interaction between lithium composition and surface reaction rate controls the kinetics and uniformity of the insertion reaction of Li ions at the solid-liquid interface, and then the rate capability and cycle life of Li-ion battery are determined.

Fig. 17. Scheme of the insertion pathway as a function of the lithiation rate[55]
In addition, Lim[52]also observed the Li com- position distribution of partial delithium samples at 1 C rate cycle1 and 12 h relaxation under theexperiment. The results suggest that the phase boundary between the Li-rich and Li-lean regions is obvious, indicating the phase separation under the influence of elastic strain dominates the Li distri- bution of the equilibrium state, namely, the Li- insertion/extraction mechanism in equilibrium state is more consistent with the “phase-boundary controlled” one.
Li[53]also employed the latestXRD to confirm when the dimension of LiFePO4in the crystal direction [100] was reduced to the width of equilibrium phase boundary (about 12 nm), and the solid solubility of Li can increase in the LiFePO4and Li1−βFePO4phases, that is, the solid solution limitation of Li increases while the miscibility gap decreases. This viewpoint also supports the “diffusion-controlled” mechanism, and confirms the miscibility gap of Li has high orientation and dependency.
In conclusion, the researches have demonstrated a variety of possibilities for the phase transition mechanisms during the Li-insertion/extraction of LiFePO4. In most cases, the improvement of material performance depends on the in-depth study of the material foundation and deep understanding of the material essence. Therefore, it has a profound significance to investigate the Li-insertion/extraction of LiFePO4mechanism.
4 PROSPECTS
The study on the mechanism of the Li-inser- tion/extraction of LiFePO4, from the initial theore- tical model to the observation and analysis oftechnique, has experienced unprecedented progress, and the experimental research is also more direct and in-depth. It should be noted that the mechanism of Li-insertion/extraction is closely relatedto many factors such as grain size of samples (nano or micro), morphology (spherical, lamellar or wire rod), the state of the sample (equilibrium or non-equilibrium), the test conditions (rate of Li-insertion/extraction), the way of Li-insertion/extraction (chemicals or electrochemical), and so on. Therefore, it does not simply determine the possible mechanism of Li-insertion/extraction, but it is necessary to combine the state and experimental conditions of the sample for analysis to reach a systematic and valuable con- clusion. For example, in non-equilibrium conditions, the samples with the nanometer scale are dominated by the “diffusion-controlled” mechanism. The samples with micron-sized LiFePO4may follow the different mechanisms at different rates of Li-inser- tion/extraction. As another example, the properties of LiFePO4may have a strong correlation with the temperature. At low temperature, lithium ions in the electrolyte slowly transfer, the impedance of surface membrane on the electrode/electrolyte interface increases, thus the charge transfer impedance on the electrode surface increases, and the diffusion of lithium ions in the electrode is slow. Therefore, the Li-insertion/extraction mechanisms of LiFePO4at low temperature may be different from the ones at normal temperature, which will be a valuable research topic in the future.
In recent years, the development ofobservation technique has brought a new opportunity for the research on the Li-insertion/extraction mechanisms of LiFePO4. This method is more intuitive, and can directly observe the extraction mode of lithium ions at the atomic level, which is the biggest progress in the research on the Li-inser- tion/extraction mechanisms. If theobservation technique is used to study systematically the samples with different grain sizes and morphology, sample states, test conditions, and Li-insertion/extraction methods, more comprehensive and profound understanding will be obtained.
At present, the theoretical research on Li-inser- tion/extraction mechanisms of LiFePO4is still inadequate. It is rare to study the mechanisms on different sample states and experimental conditions, which will become the focus of our future research.
(1) Padhi, A. K.; Nanjundaswamy, K. S.; Goodenough, J. B. Phospho-olivines as positive-electrode materials for rechargeable lithium batteries.1997, 144, 1188-1194.
(2) Ritchie, A.; Howard, W. Recent developments and likely advances in lithium-ion batteries.2006, 162, 809-812.
(3) Tarascon, J. M.; Armand, M. Issues and challenges facing rechargeable lithium batteries.,–367.
(4) Whittingham, M. S. Materials challenges facing electrical energy storage.. 2008, 33, 411–419.
(5) Ohzuku, T.; Brodd, R. J. An overview of positive-electrode materials for advanced lithium-ion batteries.. ,–456.
(6) Jugovic, D.; Uskokovic, D. A review of recent developments in the synthesis procedures of lithium iron phosphate powders.2009, 190, 538–544.
(7) Scrosati, B.; Garche, J. Lithium batteries, status, prospects and future.2010, 195, 2419–2430.
(8) Yu, F.; Zhang, L. L.; Lai, L. F. High electrochemical performance of LiFePO4cathode material viamicrowave exfoliated
graphene oxide.2015, 151, 240–248.
(9) Meethong, N.; Kao, Y. H.; Speakman, S. A.Aliovalent substitutions in olivine lithium iron phosphate and impact on structure and properties.2009, 19, 1060–1070.
(10) Fan, Q.; Lei, L.; Sun, Y.Biotemplated synthesis of LiFePO4/C matrixes for the conductive agent-free cathode of lithium ion batteries.. 2013, 244, 702–706.
(11) Yang, Z. G.; Zhang, J. L.; Kintner-Meyer, M. C. W. Electrochemical energy storage for green grid.2011, 111, 3577–3613.
(12) Tarascon, J. M.; Armand, M. Issues and challenges facing rechargeable lithium batteries.2001, 414, 359–367.
(13) Kim, S.; Zhang, Z. X.; Wang, S. L. Electrochemical and structural investigation of the mechanism of irreversibility in Li3V2(PO4)3cathodes.2016, 120, 7005–7012.
(14) Dominko, R.; Gaberscek, M.; Drofenik, J. The role of carbon black distribution in cathodes for Li ion batteries.2003, 119–121, 770–773.
(15) Chung, S. Y.; Bloking, J. T.; Chiang, Y. T. Electronically conductive phospho-olivines as lithium storage electrodes.. 2002, 1, 123–128.
(16) Yu, F.; Zhang, J.; Yang, Y. Preparation and characterization of mesoporous LiFePO4/C microsphere by spray drying assisted template method.2009, 189, 794–797.
(17) Delacourt, C.; Poizot, P.; Levasseur, S.Size effects on carbon-free LiFePO4powders, the key to superior energy density.. 2006, 9, A352–A355.
(18) Zane, D.; Carewska, M.; Scaccia, S. Factor affecting rate performance of undoped LiFePO4.2004, 49, 4259–4271.
(19) Chen, Z.; Dahn, J. R.Reducing carbon in LiFePO4/C composite electrodes to maximize specific energy, volumetric energy, and tap density.2002, 149, A1184–A1189.
(20) Kim, D. H.; Kim, J. Synthesis of LiFePO4nanoparticles in polyol medium and their electrochemical properties.2006, 9, A439–A442.
(21) Gibot, P.; Casas-Cabanas, M.; Laffont, L.Room-temperature single-phase Li insertion/extraction in nanoscale LiFePO4.2008, 7, 741–747.
(22) Ravet, N.; Goodenough, J. B.; Besner, S. Improved iron based cathode material.1999, Honolulu, 99-2HI, Abstract 127. f
(23) Gong, C. L.; Xue, Z. G.; Wen, S. Advanced carbon materials/olivine LiFePO4composites cathode for lithium ion batteries.2016, 318, 93–112.
(24) Gong, H.; Xue, H. R.; Wang, T.synthesis of monodisperse micro-nanospherical LiFePO4/carbon cathode composites for lithium-ion batteries.2016, 318, 220–227.
(25) Zhu, J. N.; Li, W. C.; Cheng, F. Li-ion battery cathode by a two-stage microwave solvothermal process.2015, 26, 13920–13925.
(26) Wang, Y. G.; Wang, Y. R.; Hosono, E. J.The design of a LiFePO4/carbon nanocomposite with a core-shell structure and its synthesis by an in situ polymerization restriction method.2008, 47, 7461–7465.
(27) Wu, X. L.; Jiang, L. Y.; Cao, F. F. LiFePO4nanoparticles embedded in a nanoporous carbon matrix, superior cathode material for electrochemical energy-storage devices.2009, 21, 2710–2714.
(28) Ma, Z.; Shao, G.; Fan, Y. Tunable morphology synthesis of LiFePO4nanoparticles as cathode materials for lithium ion batteries.2014, 6, 9236–9244.
(29) Ma, Z.; Fan, Y.; Shao, G.catalytic synthesis of high-graphitized carbon-coated LiFePO4nanoplates for superior Li-Ion battery cathodes.2015, 7, 2937–2943.
(30) Wang, G.; Ma, Z.; Shao, G. Synthesis of LiFePO4@carbon nanotube core-shell nanowires with a high-energy efficient method for superior lithium ion battery cathodes.2015, 291, 209–214.
(31) Liu, L.; Zhang, H. J.; Chen, X. Unique synthesis of sandwiched graphene@(Li0.893Fe0.036)Co(PO4) nanoparticles as high-performance cathode materials for lithium-ion batteries.2015, A3, 12320–12327.
(32) Sun, P.; Zhang, H.; Shen, K. Preparation of V-doped LiFePO4/C as the optimized cathode material for lithium ion batteries.2015, 15, 2667–2672.
(33) Andersson, A. S.; Thomas, J. O. The source of first-cycle capacity loss in LiFePO4.2001, 97-98, 498-502.
(34) Delacourt, C.; Poizot, P.; Tarascon, J. M. The existence of a temperature-driven solid solution in LiFePO4for 0≤≤1.2005, 4, 254-260.
(35) Eills, B.; Perry, K. L.; Ryan, H. D.Small polaron hopping in LiFePO4solid solutions, coupled lithium-ion and electron mobility.2006, 128, 11416–11422.
(36) Malik, R.; Zhou, F.; Ceder, G. Kinetics of non-equilibrium lithium incorporation in LiFePO4.2011, 10, 587-590.
(37) Jones, L. A. R.. Oxford University Press 2002.
(38) Delmas, C.; Maccario, M.; Croguennec, L. Lithium deintercalation in LiFePO4nanoparticles via a domino-cascade model.2008, 7, 665-671.
(39) Laffont, L.; Delacourt, C.; Gibot, P. Study of the LiFePO4/FePO4two-phase system by high-resolution electron energy loss spectroscopy.2006, 18, 5520-5529.
(40) Ramana, C. V.; Mauger, A.; Gendron, F. Study of the Li-insertion/extraction process in LiFePO4/FePO4.2009, 187, 555–564.
(41) Allen, J. L.; Jow, R. T.; Wolfenstine, J. Kinetic study of the electrochemical FePO4to LiFePO4phase transition.2007, 19, 2108-2111.
(42) Dedryvère, R.; Maccario, M.; Croguennec, L.X-ray photoelectron spectroscopy investigations of carbon-coated LiFePO4materials.2008, 20, 7164-7170.
(43) Bai, P.; Cogswell, D. A.; Bazant, M. Z. Suppression of phase separation in LiFePO4nanoparticles during battery discharge.2011, 11, 4890–4896.
(44) Oyama, G.; Yamada, Y.; Natsui, R. I. Kinetics of nucleation and growth in two-phase electrochemical reaction of LiFePO4.2012, 116, 7306–7311.
(45) Gu, L.; Zhu, C. B.; Li, H. Direct observation of lithium staging in partially delithiated LiFePO4at atomic resolution.2011, 133, 4661-4663.
(46) Chang, H. H.; Chang, C. C.; Wu, H. C. Study on dynamics of structural transformation during charge/discharge of LiFePO4cathode2008, 10, 335-339.
(47) Wang, X. J.; Jaye, C.; Nam, K. W. Investigation of the structural changes in Li1-xFePO4upon charging by synchrotron radiation techniques.2011, 21, 11406-11411.
(48) Wang, J. J.; Chen-Wiegart, Y. C. K.; Wang, J. In-operando tracking phase transformation evolution of lithium iron phosphate with hard X-ray microscopy.2014, 5, 4570.
(49) Sharma, N.; Guo, X. W.; Du, G. D.Direct evidence of concurrent solid-solution and two-phase reactions and the nonequilibrium structural evolution of LiFePO4.2012, 134, 7867-7873.
(50) Yu, X. Q.; Wang, Q.; Zhou, Y. N. High rate delithiation behaviour of LiFePO4studied by quick X-ray absorption spectroscopy.2012, 48, 11537-11539.
(51) Niu, J. J.; Kushima, A.; Qian, X. F.observation of random solid solution zone in LiFePO4electrode.2014, 14, 4005-4010.
(52) Lim, J.; Li, Y. Y.; Alsem, D. H. Origin and hysteresis of lithium compositional spatiodynamics within battery primary particles2016, 353, 566-571.
(53) Li, Z. J.; Yang, J. X.; Li, C. J. Orientation-dependent lithium miscibility gap in LiFePO4.2018, 30, 3, 874-878.
7 March 2018;
26 September 2018
① This work was supported by the National Natural Science Foundation of China (No. 51504196) and Key Research and Development Plan of Shaanxi Province (No. 2017ZDXM-GY-039)
. E-mail: wyf7777v@126.com
10.14102/j.cnki.0254-5861.2011-2000

- 结构化学的其它文章
- Nanoclusters Au19Pd and Au19Pt Catalyzing CO Oxidation: a Density Functional Study①
- Synthesis, Single-crystal Structure and Fluorescence Property of a New Boron Compound Based on 2-(2΄-Hydroxyphenyl)-1H-benzimidazole①
- Synthesis of a NovelImidazole Ionic Liquid Modified Mesoporous Silica SBA15 for Selective Separation and Determination of Inorganic Arsenic in Rice①
- Theoretical Investigations on the Structural and Electronic Properties of WO3Polymorphs①
- Studies on a Series of Coumarin Derivatives for Anticancer Activity against Breast Carcinoma Cell Line MCF-7 and Their Molecular Design
- Surface Modification of Titanium Oxide by Indium for Efficient Photocatalytic Hydrogen Generation①