
Theoretical Study on the Reaction Mechanism of o-Aminophenol, Acetic Acid and Phosphorus Oxytrichloride One-pot to Form 2-Methyl Benzoxazole①
2019-01-05 09:34ZHANGFuLan
结构化学 2018年12期
ZHANG Fu-Lan
Theoretical Study on the Reaction Mechanism of-Aminophenol, Acetic Acid and Phosphorus Oxytrichloride One-pot to Form 2-Methyl Benzoxazole①
ZHANG Fu-Lan②
(408003)
The reaction mechanism of-aminophenol, acetic acid and phosphorus oxytrichlo- ride in one-pot to form 2-methyl benzoxazole was studied by density functional theory. The geometries of the reactants, transition states, intermediates and products were optimized at the GGA/PW91/DNP level. Vibration analysis was carried out to confirm the transition state structure. Two possible reaction pathways were investigated in this study. The result indicates that the reaction Re→TS1→IM1→TSA2→IMA2→TSA3→IMA3→TSA4→IMA4→TSA5→P2is the main pathway, the activation energy of which is the lowest. Re→TS1→IM1 is the rate-limiting step, with the activation energy being 221.54 kJ·mol-1and the reaction heat being 10.06 kJ·mol-1. The dominant product predicted theoretically is in agreement with the experiment results.
-aminophenol, acetic acid, phosphorus oxytrichloride, 2-methyl benzoxazole, density functional, reaction mechanism;
1 INTRODUCTION
Heterocyclic compounds exist widely in nature. It is the largest number of organic compounds. Benzo- xazoleis a kind of nitrogenous heterocyclic compounds which have been widely used. It is important for pilot skeleton and maternal in organic synthesis. Study on the synthesis of these com- pounds is one of the focuses in recent twenty years, because it has many physiological activities, drug activities, and so on[1, 2]. For example, they have been widely applied by antimicrobial[3], anticon- vulsant[4], antifungal[5], anti-inflammation[6], antitu- mor[7, 8], and so forth. According to relevant research, benzoxazoles were used as heat-resistant materials, because they have high heat resistance and corrosion resistance[9]. They have been widely applied by fluorescent brightener and scintillator[10, 11].
In recent years, benzoxazoles are so widely used, and their synthesis reaction has become attractive in chemistry. There are many ways in the high-yield synthesis of benzoxazoles[12-17]. In particular, Tang. developed a new way[17], as shown in Fig. 1. Four new series of 2-alkyl, 2-aryl and 2-styryl benzoxazoles were synthesized by using-amino- phenol, acetic acid and phosphorus oxytrichloride in refluxing CHCl3in one-pot. Compared with the traditional methods, this method has the advantages of mild reaction conditions, simple operation, good yields, and easily available reaction substrate.However, the mechanism of these reactions still remains unclear. To make a better understanding of these reactions, we investigated the typical reaction mechanism of-aminophenol, acetic acid and phosphorus oxytrichloride one-pot to form 2-methyl benzoxazole by density functional theory (DFT). I hope that the research can provide a theoretical base for the synthesis of benzoxazoles. The computa- tional details are described in the next section. In section 3, we present the calculated results and discuss the reaction mechanism, followed by a conclusion in section 4.

Fig. 1. Synthesis of 2-methyl benzoxazole
2 CALCULATION METHODS
All calculations have been performed using Dmol3code[18, 19]as implemented in Accelrys Materials Studio 5.0. The generalized gradient approximation (GGA) with the Perdew-Wang (PW91)[20]exchange-correlation functional is selected in the DFT calculations. All electrons are computationally inexpensive with good approxima- tion for elements with atomic numbers less than 21. The convergence criteria for geometry optimization are 2×10-5hartree, 0.004 hartree/Å, 0.005 Å, and 1 × 10-5hartree for the energy, force, displacement, and selfconsistent field (SCF) density, respectively. Dmol3utilizes a basis set of numeric atomic func- tions, which are exact solutions to the Kohn-Sham equations for the atom[21]. The basis set of double numerical plus polarization (DNP) is used throughout the study.
Preliminary transition state geometries are obtained using the integrated linear synchronous transit/quadratic synchronous transit (LST/QST) method[22]. All structures identified as stationary points are subject to full-frequency analysis to verify their classification as equilibrium geometries (zero imaginary frequencies) or transition states (one imaginary frequency). The solvent effects of species have also been acquired by COMSO.
3 RESULTS AND DISCUSSION
In this work, we have explored the reaction of acetic acid (Re1), phosphorus oxytrichloride (Re2), and-aminophenol (Re3), as shown in Fig. 2.The total energies, relative energies and frequencies of different compounds are listed in Table 1. The corresponding geometries of the reactants and products are shown in Fig. 3. The corresponding geometries of the intermediates and transition states are shown in Figs. 4 and 5, respectively. Thediagram of relative energies along the channels of reactions is shown in Fig. 6.
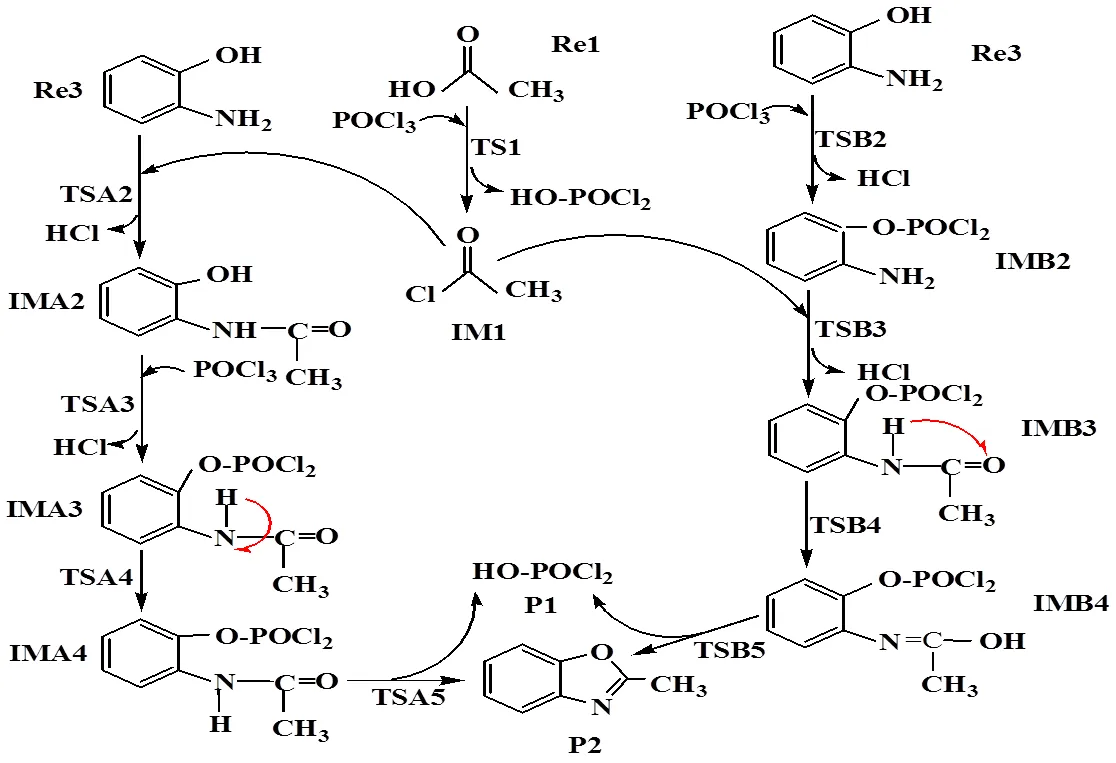
Fig. 2. Processes for the synthesis of 2-methyl benzoxazole
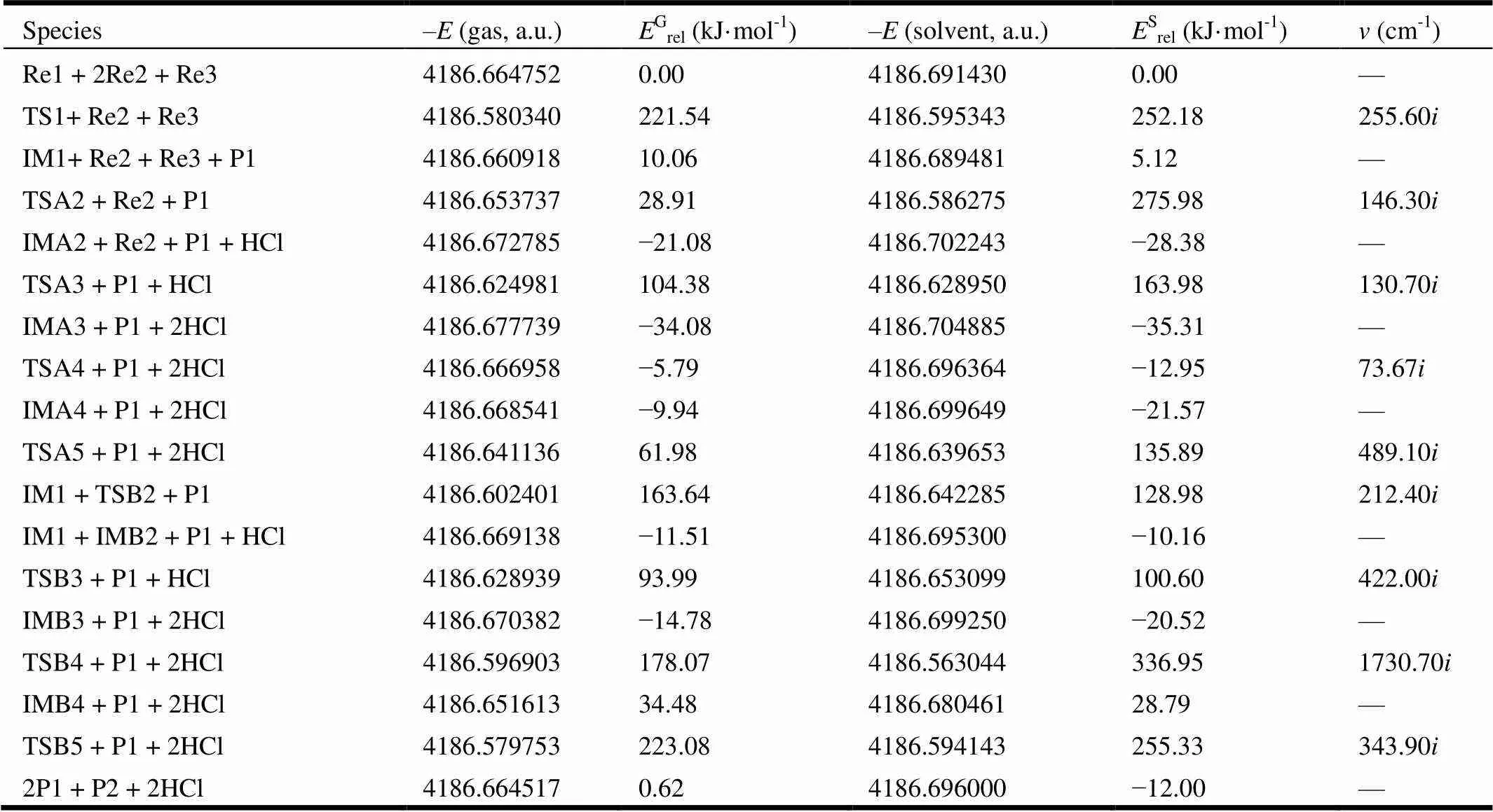
Table 1. Total Energies (E(a.u.)), Relative Energies (Erel/(kJ·mol-1))–and Frequencies ν (cm-1) of the Stationary Points on the Reaction Paths
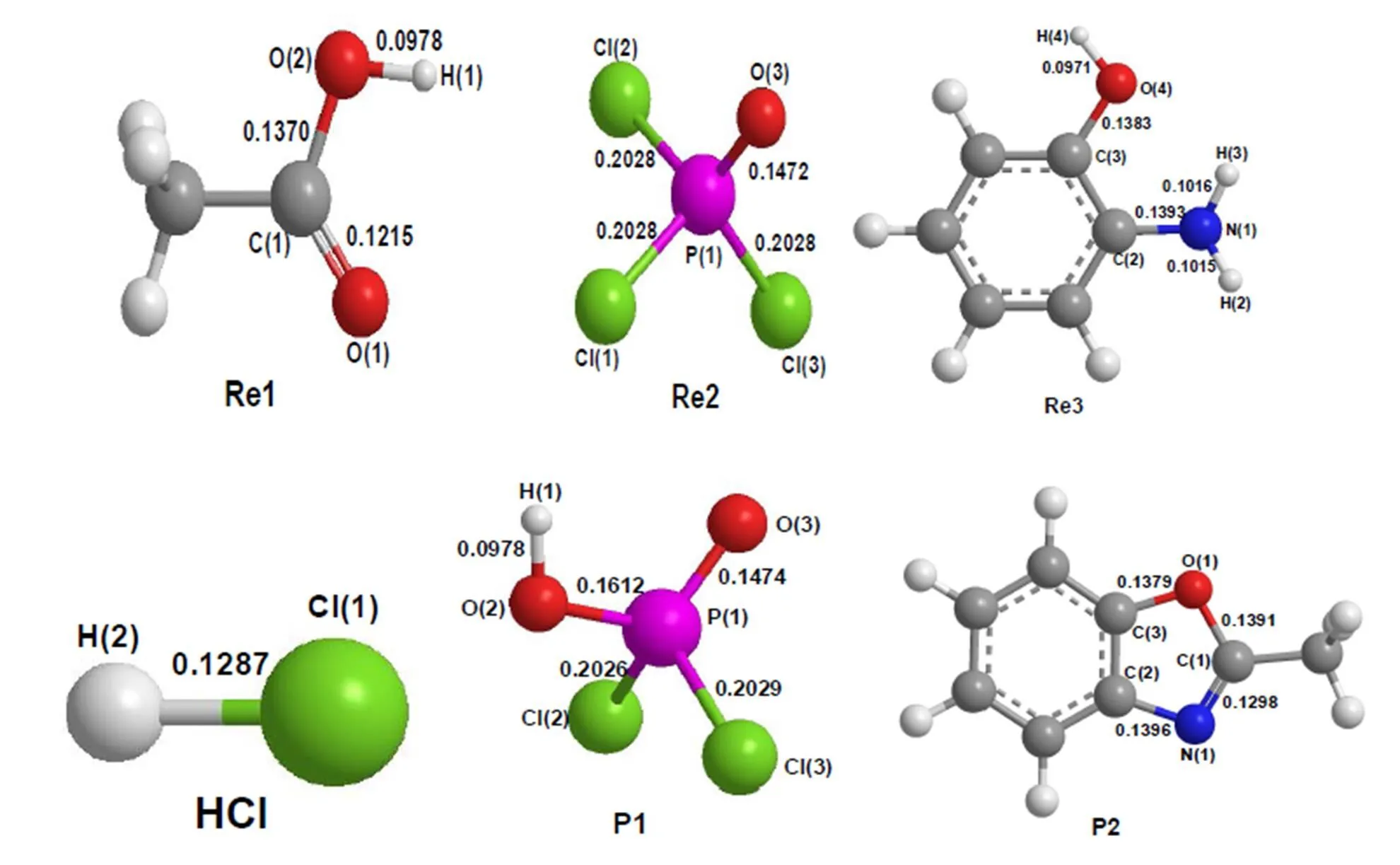
Fig. 3. Geometric parameters of the reactants and products (Bond length in nm and bond angle in degree)
3. 1 Reaction mechanism analysis
The reaction mechanism ofacetic acid (Re1),phosphorus oxytrichloride (Re2), and-amino- phenol (Re3) one-pot to form 2-methyl benzoxazole was studied by density functional theory at the GGA/PW91/DNP level. The first stage is that acetic acid and phosphorus oxytrichloride formed IM1, which is the substitution reaction. In the step, the hysroxyl bond of Re1 is replaced by the Cl(1) atom of Re2. Through a ring transition state TS1, IM1 and P1 are prepared. In this process, the activation energy is 221.54 kJ·mol-1, the heat of reaction is 10.06 kJ·mol-1and the only imaginary frequency is 255.60cm-1, as shown in Table 1.
The C(1)–O(2) bonds are 0.1370 and 0.1958 nm in Re1 and TS1, respectively. The P(1)–Cl(1) bonds are 0.2028 and 0.2428 nm in Re2 and TS1, respectively. The C(1)–Cl(1) bonds are 0.2230 and 0.1829 nm in TS1 and IM1, respectively. The O(2)–P(1) bonds are 0.1814 and 0.1612 nm in TS1 and P1, respectively. The bond lengths of C(1)–O(2) and P(1)–Cl(1) are increased by 0.0588 and 0.0300 nm, while those of C(1)–Cl(1) and O(2)–P(1) are decreased by 0.0401 and 0.0202 nm, respectively. Obviously, the C(1)–O(2) and P(1)–Cl(1) bonds are partly broken, and the C(1)–Cl(1) and O(2)–P(1) bonds are partly formed in TS1. After the transition state TS1, intermediate IM1 and product P1 are formed.
In the second stage, there are two possible reac- tion pathways from intermediate IM1 to the products P1 and P2: in paths A and B. The two reaction pathways are discussed as follows.
3. 1. 1 Analysis of the chemical reaction mechanism of path A
In path A,-aminophenol (Re3) form P2, in order of three stages of acylation, nucleophilic addition, and cyclization. Our calculations indicate that the whole reaction process consists of four steps, during which three intermediates and four transition states are formed.
Firstly, with acylation reaction of compound IM1 and-aminophenol (Re3), compound IMA2 was prepared. In the process of forming IMA2, the NH2of Re3 reacts with the C(1)–Cl(1) of IM1. The H atom of NH2transfers to Cl(1), while the NH of NH2transfers to C(1). The acylation reaction occurs easily because the activation energy for the reaction from complex IM1 to TSA2 is 18.85 kJ·mol-1. As listed in Table 1, the heat of reaction is –30.14 kJ·mol-1and the only imaginary frequency is 146.30i cm-1. The N(1)–H(2) bonds are 0.1015 and 0.1419 nm in Re3 and TSA2, respectively. The C(1)–Cl(1) bonds are 0.1829 and 0.2477 nm in IM1 and TSA2, respectively. The C(1)–N(1) bonds are 0.2901 and 0.1382 nm in TSA2 and IMA2, respec- tively. The bond lengths of N(1)–H(2) and C(1)–Cl(1) are increased by 0.0404 and 0.0648 nm, while that of C(1)–N(1) is decreased by 0.1519 nm, respectively. Obviously, the N(1)–H(2) and C(1)– Cl(1) bonds are partly broken, and the C(1)–N(1) bond is partly formed in TSA2. After the reaction surpasses the transition state TSA2, the interme- diates IMA2 and HClare formed.
Secondly, with nucleophilic addition reaction of compound IMA2 and phosphorus oxytrichloride (Re2), compound IMA3 was prepared. In the process of forming IMA3, the P(1) of phosphorus oxytrichloride transfers to the O(4) atom of OH in IMA2, while the H(4) atom of OH in IMA2 transfers to the Cl(1) of Re2. As listed in Table 1, the activation energy is 125.46 kJ·mol-1for the reaction from complex IMA2 to TSA3. To verify the process, we have located the ring transition state TSA3 (Fig. 5). For the TSA3 structure, the only imaginary frequency is 130.70·cm-1(Table 1). The analysis on the vibration modes indicates that this imaginary frequency is associated with Cl(1)–H(4) and P(1)–O(4) stretching motions. As listed in Figs. 3, 4, and 5, the O(4)–H(4) bonds are 0.0970 and 0.2266 nm in IMA2 and TSA3, respectively. The P(1)–Cl(1) bonds are 0.2028 and 0.2994 nm in Re2 and TSA3, respectively. The O(4)–P(1) bonds are 0.3450 and 0.1620 nm in TSA3 and IMA3, respectively. The bond lengths of O(4)–H(4) and P(1)–Cl(1) are increased, while that of O(4)–P(1) is decreased, respectively. Obviously, the O(4)–H(4) and P(1)–Cl(1) bonds are partly broken, and the O(4)–P(1) bond is partly formed in TSA3. After the transition state TSA3, and intermediate IMA3 and HClare formed.
Subsequently, the IMA3 is isomerized. Rotating aroundC(2)–N(1) bond,IMA4 is prepared through transition state TSA4. In this process, the activation energy is 28.29 kJ·mol-1, the heat of reaction is 24.14 kJ·mol-1, and the only imaginary frequency is 73.67cm-1, as shown in Table 1.
Finally, the IMA4 is isomerized. The imino H(3) atom transfers to the phenolic hydroxyl O(4) atom, and carbonyl O(1) crashes to C(3) on the benzene ring. Through a six-membered ring transition state TSA5, the main product P2 and by-product P1 are prepared. In this process, the activation energy is 71.92 kJ·mol-1, the heat of reaction is 67.77 kJ·mol-1, and the only imaginary frequency is 489.10cm-1, as shown in Table 1. The C(3)–O(4) bonds are 0.1406 and 0.2394 nm in IMA4 and TSA5, respectively. The N(1)–H(3) bonds are 0.1015 and 0.1030 nm in IMA4 and TSA5, respectively. The O(4)–H(3) bonds are 0.3098 and 0.0978 nm in TSA3 and P1, respectively. The C(3)–O(1) bonds are 0.2377 and 0.1379 nm in TSA5 and P2, respectively. The C(1)–N(1) bonds are 0.1392, 0.1311, and 1298 nm in IMA4, TSA5, and P2, respectively. The C(1)–O(1) bonds are 0.1222, 0.1237, and 1391 nm in IMA4, TSA5, and P2, respectively. The bond lengths of N(1)–H(3), C(3)–O(4), and C(1)–O(1) are increased, while those of C(3)–O(1), O(4)–H(3), and C(1)–N(1) are decreased, respectively. Obviously, the N(1)– H(3), C(3)–O(4), and C(1)–O(1) bonds are partly broken, and the C(3)–O(1), O(4)–H(3), and C(1)– N(1) bonds are partly formed in TSA5.
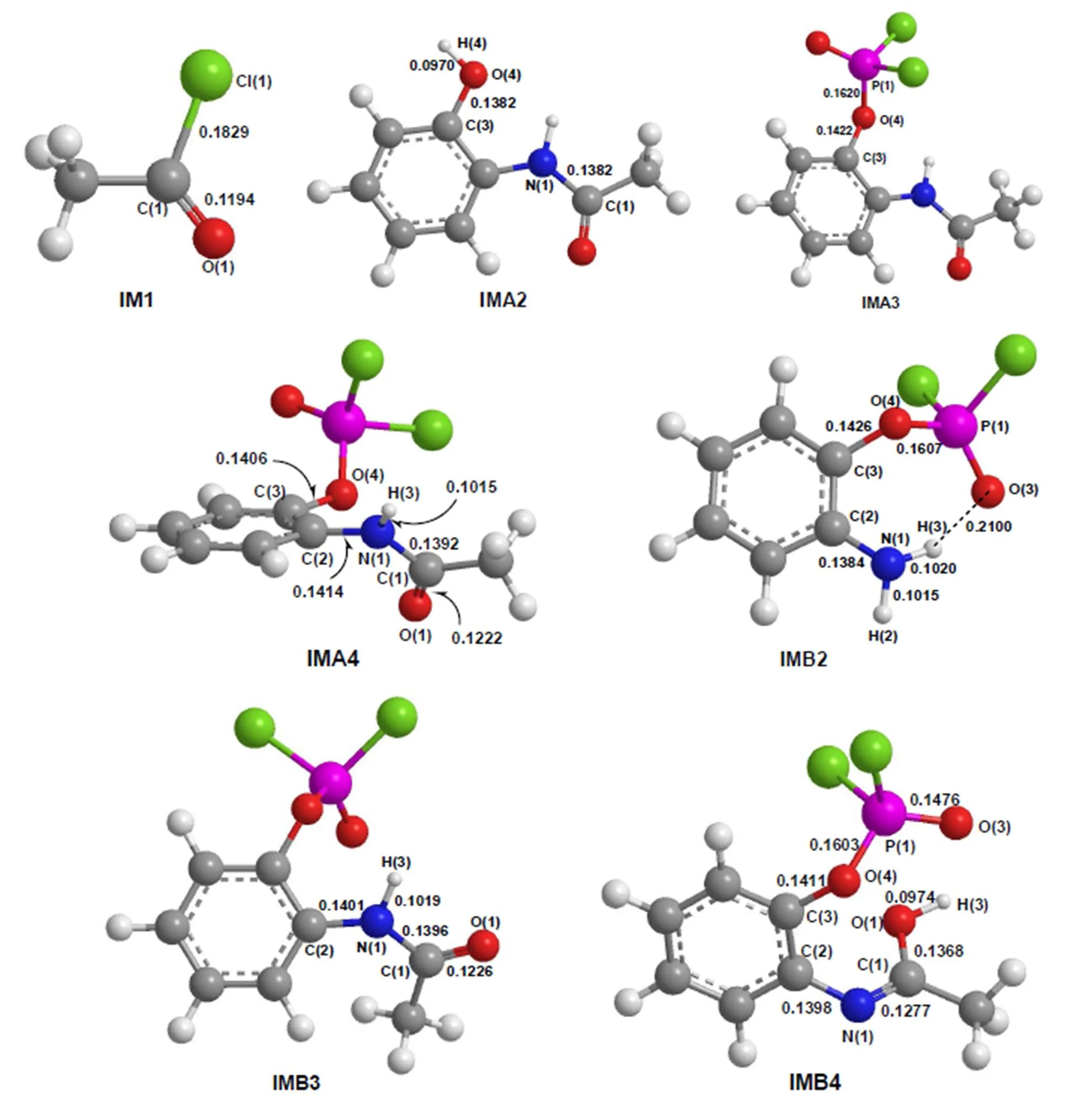
Fig. 4. Geometric parameters of the intermediates (Bond length in nm)
Fig. 5. Geometric parameters of the transition states (Bond length in nm)
3. 1. 2 Analysis of chemical reaction mechanism of path B
In path B,-aminophenol (Re3) forms P2, in order of three stages of nucleophilic addition, acylation, and cyclization. Similarly, in path A, our calculations indicate that the whole reaction process consists of four steps, during which three interme- diates and four transition states are formed, too.
Firstly, similar to forming IMA3 in path A, with nucleophilic addition reaction of-aminophenol (Re3) and phosphorus oxytrichloride (Re2), com- pound IMB2 was prepared. In the process of forming IMB2, the P(1) of phosphorus oxytri- chloride transfers to the O(4) atom of OH in-aminophenol (Re3), while the H(4) atom of OH in-aminophenol (Re3) transfers to the Cl(1) of Re2. As listed in Table 1, the activation energy is 153.58 kJ·mol-1for the reaction. To verify the process, we have located the ring transition state TSB2 (Fig. 5). For the TSB2 structure, the only imaginary frequency is 212.40cm-1(Table 1). The analysis on the vibration modes indicates that this imaginary frequency is associated with Cl(1)–H(4) and P(1)–O(4) stretching motions. The O(4)–H(4) bonds are 0.0971 and 0.1472 nm in Re3 and TSB2, respectively. The P(1)–Cl(1) bonds are 0.2028 and 0.2965 nm in Re2 and TSB2, respectively. The O(4)–P(1) bonds are 0.2854 and 0.1607 nm in TSB2 and IMB2, respectively. The bond lengths of O(4)–H(4) and P(1)–Cl(1) are increased, while that of O(4)–P(1) is decreased, respectively. Obviously, the O(4)–H(4) and P(1)–Cl(1) bonds are partly broken, and the O(4)–P(1) bond is partly formed in TSB2. After the transition state TSB2, the intermediates IMB2 and HClare formed. Compared with path A, the bond length of O(4)–P(1) is much shorter because there is a hydrogen bond between O(3) of phosphorus oxytrichloride and amino H(3) of-aminophenol.
Secondly, similar to forming IMA2 in path A, with the acylation reaction of compounds IM1 and IMB2, compound IMB3 was prepared. In the process of forming IMB3, the NH2of IMB2 reacts with the C(1)–Cl(1) of IM1. The H atom of NH2transfers to Cl(1), while the NH of NH2transfers to C(1). The acylation reaction occurs easily because the activation energy for the reaction from complex IMB3 to TSB3 is 105.50 kJ·mol-1. As listed in Table 1, the heat of reaction is –3.27 kJ·mol-1, and the only imaginary frequency is 422.00cm-1. The N(1)–H(2) bonds are 0.1015 and 0.1067 nm in IMB2 and TSB3, respectively. The C(1)–Cl(1) bonds are 0.1829 and 0.2481 nm in IM1 and TSB3, respectively. The C(1)–N(1) bonds are 0.1898 and 0.1396 nm in TSB3 and IMB3, respectively. The bond lengths of N(1)– H(2) and C(1)–Cl(1) are increased by 0.0052 and 0.0652 nm, while that of C(1)–N(1) is decreased by 0.0502 nm, respectively. Obviously, the N(1)–H(2) and C(1)–Cl(1) bonds are partly broken, and the C(1)–N(1) bond is partly formed in TSB3. After the transition state TSB3, intermediates IMB3 and HClare formed.
Subsequently,the IMB3 is isomerized, and the imino proton H(3) transfers to the carbonyl O(1), thus papering compound IMB4 through a four- membered ring transition state TSB4. In this process, the activation energy is 192.85 kJ·mol-1,the heat of reaction is 49.26 kJ·mol-1and the only imaginary frequency is 1730.70cm-1, as shown in Table 1. The C(1)–O(1) bonds are 0.1226, 0.1294, and 0.1368 nm in IMB3, TSB4, and IMB4, respectively. The C(1)–N(1) bonds are 0.1396, 0.1327, and 0.1277 nm in IMB3, TSB4, and IMB4, respectively. The N(1)–H(3) bonds are 0.1019 and 0.1335 nm in IMB3 and TSB4, respectively. The O(1)–H(3) bonds are 0.1332 and 0.0974 nm in TSB4 and IMB4, respectively. The bond lengths of N(1)–H(3) and C(1)–O(1) are increased, while those of C(1)–N(1) and O(1)–H(3) are decreased, respectively. Meanwhile, the C(1)=N(1) is formed.
Finally, the IMB4 is isomerized, the hydroxy H(3) atom transfers to the phenolic hydroxyl O(3) atom, and the hydroxy O(1) crashes to C(3) on the benzene ring. Through a six-membered ring transition state TSB5, the main product P2 and the by-product P1 are prepared. In this process, the activation energy is 188.60 kJ·mol-1,the heat of reaction is –33.86 kJ·mol-1, and the only imaginary frequency is 343.90cm-1, as shown in Table 1. The C(3)–O(4) bonds are 0.1411 and 0.2114 nm in IMB4 and TSB5, respectively. The O(1)–H(3) bonds are 0.0974 and 0.1091 nm in IMB4 and TSB5, respectively. The O(3)–H(3) bonds are 0.1384 and 0.0978 nm in TSB5 and P1, respectively. The C(3)–O(1) bonds are 0.2146 and 0.1379 nm in TSB5 and P2, respectively. The bond lengths of O(1)–H(3) and C(3)–O(4) are increased, while those of C(3)–O(1) and O(3)–H(3) are decreased, respectively.
The configuration parameters of the reaction processes are shown in Figs. 3, 4 and 5, respectively.
3. 2 Energy analysis
As shown in Fig. 6, the microcosmic reaction mechanism of-aminophenol, acetic acid and phosphorus oxytrichloride has two possible reaction pathways in the gas phase.The first stage is that acetic acid and phosphorus oxytrichloride formed IM1, which is the substitution reaction. In this process, the formation of intermediate IM1 is so difficult because the activation energy for the reaction from Re to TS1 is 221.54 kJ·mol-1, and Re→TS1→IM1 is the rate-limiting step in the whole reaction process. Subsequently, the results show the two possible reaction pathways from the interme- diate IM1 to the 2-methyl benzoxazole. In path A, IMA2→TSA3→IMA3 is the rate-limiting step and the activation energy is 125.46 kJ·mol-1. In path B, IMB3→TSB4→IMB4 is the rate-limiting step and the activation energy is 192.85 kJ·mol-1. By contrast, it is better to choose path A in the course of forming P2. On the other hand, in the salvation of CHCl3,IM1→TSA2→IMA2 is the rate-limiting step and the activation energy is 270.86 kJ·mol-1in path A, while IMB3→TSB4→IMB4 is the rate-limiting step and the activation energy is 357.47 kJ·mol-1in path B. Path A is also the superior path in these two paths.
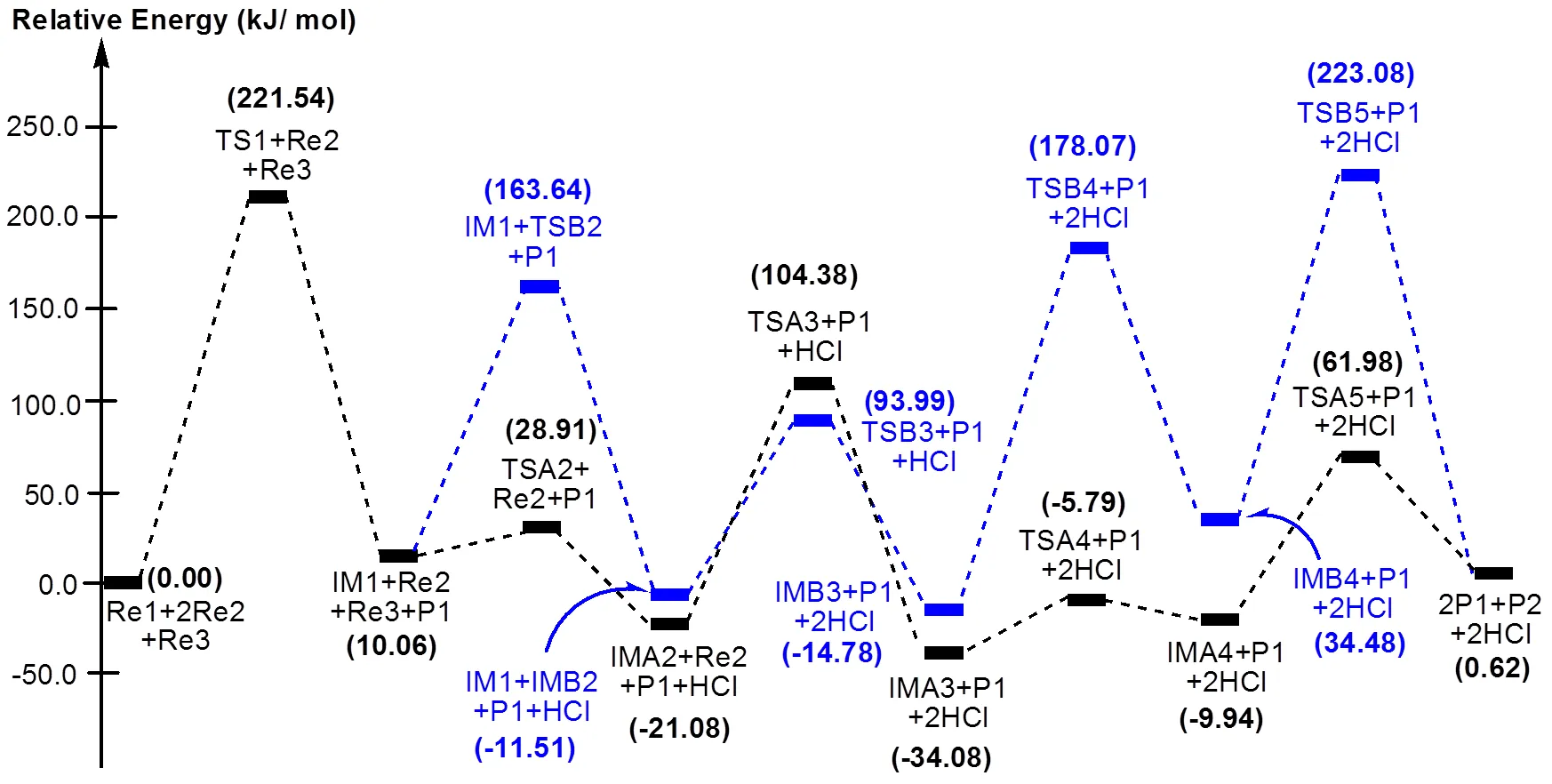
Fig. 6. Diagram of relative energies along the channels of the reactions
Through the preceding analyses we can conclude that the main pathway of the microcosmic reaction mechanism of-aminophenol, acetic acid and phos- phorus oxytrichloride is Re→TS1→IM1→TSA2→ IMA2→TSA3→IMA3→TSA4→IMA4→TSA5→ P2. The activation energy of Re→TS1→IM1, the rate-control step, in the pathway is lower by two feasible reaction pathways. The energy barrier and the heat of formation of the rate-limiting stepare 221.54 and 10.06 kJ∙mol-1, respectively. The P2 is the main product of this reaction, which is in good accordance with the experiment[17].
4 CONCLUSION
The microcosmic reaction mechanism of-amino- phenol, acetic acid and phosphorus oxytrichloride has been investigated in refluxing CHCl3in one-pot to form 2-methyl benzoxazole by density functional theory (DFT) at the GGA/PW91/DNP level. We optimize the geometric configurations of reactants, intermediates, transition states, and products. The energy analysis calculation approves the authenticity of intermediates and transition states. According to our calculations we found two feasible reaction pathways. The main pathway of the reaction is Re→TS1→IM1→TSA2→IMA2→TSA3→IMA3→TSA4→IMA4→TSA5→P2. The rate-limiting step is Re→TS1→IM1, for which the energy barrier and the heat of formation of are 221.54 and 10.06 kJ∙mol-1, respectively. The energy barrier of the rate-limiting step is higher, so this reaction must be heated, which is in agreement with the experimental conditions under microwave irradiation[17]. The dominant product predicted theoretically is 2-methyl benzoxazole, which is in agreement with the experi- mental results in reference[17].
(1) Seenaiah, D.; Reddy, P. R.; Reddy, G. M.; Padmaja, A.; Padmavathi, V.; Siva Krishna, N. Synthesis, antimicrobial and cytotoxic activities of pyrimidinyl benzoxazole, benzothiazole and benzimidazole.2014, 77, 1-7.
(2) Kaur, A.; Wakode, S.; Pathak, D. P. Benzoxazole: the molecule of diverse pharmacological importance.2015, 7, 16-23.
(3) Temiz-Arpaci, O.; Aki-Sener, E.; Yalçin, I.; Altanlar, N. Synthesis and antimicrobial activity of some 2-[-substituted-phenyl]benzoxazol- 5-yl-arylcarboxyamides.() 2002, 335, 283-288.
(4) Tan, Y. D.; He, X. Y.; Rao, B. Q.; Cheng, B. B.; Song, M. X.; Deng, X. Q. Synthesis and evaluation of the anticonvulsant activities of triazole-containing benzo[]oxazoles.2016, 36, 2449-2455.
(5) Modiya, P. R.; Patel, C. N. Synthesis and screening of antibacterial and antifungal activity of 5-chloro-1,3-benzoxazol-2(3h)-one derivatives.. 2012, 2, 29-38.
(6) Iyer, V. B.; Gurupadayya, B. M.; Sairam, K. V.; Inturi, B.; Chandan, R. S.; Tengli, A. K. Anti-inflammatory activity of 1,3,4-oxadiazoles derived from benzoxazole.2015, 2, 233-241.
(7) Paramashivappa, R.; Phani Kumar, P.; Subba Rao, P. V.; Srinivasa Rao, A.Design, synthesis and biological evaluation of benzimidazole/benzothiazole and benzoxazole derivatives as cyclooxygenase inhibitors.2003, 13, 657-660.
(8) Wang, J.; Zhang, L.; Yao, Q. Z. Synthesis and anti-tumor activities of novel pyrazole derivatives containing 1,3,4-oxadiazole.2014, 22, 730-733.
(9) Wee, D.; Yoo, S.; Kang, Y. H.; King, Y. H.; Ka, J. W.; Cho, S. Y.; Lee, C.; Ryu, J.; Yi, M. Y.; Jang, K. S.Poly(imide-benzoxazole)gate insulators with high thermal resistance for solution processed flexible indium-zinc oxide thin-film transistors.2014, 2, 6395-6401.
(10) Dick, P. F.; Coelho, F. L.; Rodembusch, F. S.; Campo, L. F. Amphiphilic ESIPT benzoxazole derivatives as prospective fluorescent membrane probes.2014, 55, 3024-3029.
(11) Ge, G. Z. Synthesis of bis(2-benzoxazolyl) ethylene series fluorescent brighteners.2017, 54, 24-28.
(12) Xiao, L. W.; Gao, H. J.; Kong, J.; Liu, G. X.; Peng, X. X.; Wang, S. J. Progress in the synthesis of 2-substituted benzoxazoles derivatives.2014, 34, 1048-1060.
(13) Sharma, H.; Sing, N.; Jang, D. O. A ball-milling strategy for the synthesis of benzothiazole, benzimidazole and benzoxazole derivatives under solvent-free conditions.2014, 16, 4922-4930.
(14) Anand, M.; Ranjitha, A.; Himaja, M. Silica sulfuric acid catalyzed microwave-assisted synthesis of substituted benzoxazoles and their antimicrobial activity.2011, 2, 211-213.
(15) Endo, Y.; Backvall, J. E. Biomimetic oxidative coupling of benzylamines and 2-aminophenols: synthesis of enzoxazoles.2012, 18, 13609-13613.
(16) Mao, Z. F.; Wang, Z.; Xu, Z. Q.; Huang, F.; Yu, Z. K.; Wang, R. Copper(II)-mediated dehydrogenative cross-coupling of heteroarenes.2012, 14, 3854-3857.
(17) Tang, Y. L.; Du, Z. B.; Jiang, G. F. A one-pot synthetic route to substituted benzoxazoles.() 2016, 47, 408-413.
(18) Delley, B. An all-electron numerical method for solving the local density functional for polyatomic molecules.1990, 92, 508-517.
(19) Delley, B. From molecules to solids with the DMol3approach.2000, 113, 7756-7764.
(20) Perdew, J. P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy.1992, 45, 13244-13249.
(21) Delley, B. Modern density functional theory: a tool for chemistry, in: theoretical and computational chemistry, Eds.: Seminario, J. M.; Politzer, P. Elsevier, Amsterdam 1995, 2.
(22) Halgren, T. A.; Lipscomb, W. N. The synchronous-transit method for determining reaction pathways and locating molecular transition states.. 1977, 49, 225-232.
3 April 2018;
13 August 2018
the Scientific and Technological Research Program of Chongqing Municipal Education Commission (KJ1601215) and the Ministry of Education “Chunhui Plan” (Z2016177)
. Born in 1976, lecture, majoring in quantum chemistry. E-mail: cjsy0606@163.com
10.14102/j.cnki.0254-5861.2011-2032

- 结构化学的其它文章
- Nanoclusters Au19Pd and Au19Pt Catalyzing CO Oxidation: a Density Functional Study①
- Synthesis, Single-crystal Structure and Fluorescence Property of a New Boron Compound Based on 2-(2΄-Hydroxyphenyl)-1H-benzimidazole①
- Review on Li-insertion/extraction Mechanisms of LiFePO4 Cathode Materials①
- Synthesis of a NovelImidazole Ionic Liquid Modified Mesoporous Silica SBA15 for Selective Separation and Determination of Inorganic Arsenic in Rice①
- Theoretical Investigations on the Structural and Electronic Properties of WO3Polymorphs①
- Studies on a Series of Coumarin Derivatives for Anticancer Activity against Breast Carcinoma Cell Line MCF-7 and Their Molecular Design