
预氧化处理时间对C/C复合材料及其SiC抗氧化涂层的影响
2019-01-05 03:30王宇杰张明瑜苏哲安童恺黄启忠
粉末冶金材料科学与工程 2018年6期
王宇杰,张明瑜,苏哲安,童恺,黄启忠
预氧化处理时间对C/C复合材料及其SiC抗氧化涂层的影响
王宇杰,张明瑜,苏哲安,童恺,黄启忠
(中南大学 粉末冶金国家重点实验室,长沙 410083)
采用基体预氧化处理结合化学气相沉积法在C/C复合材料表面制备具有嵌入式界面的SiC涂层,研究预氧化处理时间对C/C复合材料及其SiC涂层微观结构的影响,并对涂层试样进行1 500 ℃高温抗氧化性能测试。结果表明,C/C复合材料预氧化后表面呈多孔结构,基体炭的氧化速率大于炭纤维;随预氧化时间延长,C/C复合材料表面粗糙度增大,SiC涂层对基体材料的嵌入深度增加,涂层表面裂纹变窄。1 500 ℃高温氧化实验中,氧化21 h后,预氧化处理6 min和预氧化处理9 min制备的涂层试样的质量损失率分别为2.34%和1.89%;氧化26 h后,预氧化6 min的涂层表现出更好的抗氧化性能,质量损失率为4.11%。这2种试样在高温氧化21 h前涂层对基体均能产生较好的“钉扎效应”,然而26 h后,预氧化9 min的涂层试样中层片状热解炭受热应力破坏,导致界面结合强度降低,涂层迅速失效。
C/C复合材料;预氧化处理时间;SiC涂层;抗氧化性能;钉扎效应;层片状热解炭
C/C复合材料具有诸多优异的高温性能,如耐烧蚀、抗热震以及良好的高温强度保持率,是目前极少数能服役于高于2 000 ℃环境的轻质耐高温结构材料之一,广泛应用于国防军工及航空航天工业[1−3]。然而,研究表明[4−5],当温度达到450 ℃时,C/C复合材料在氧化气氛中易氧化,且随温度升高其氧化速率迅速增大,极大限制了其在高温结构材料领域的应用。因此,对C/C复合材料进行高温抗氧化防护研究十分必 要[6]。目前C/C复合材料的氧化防护技术主要分为基体改性和表面涂层两大类[7−8]。相比于基体改性,表面涂层能对C/C复合材料提供更长时间、更高温度的氧化防护,是目前最有效直接的防护途径。SiC的高温抗氧化性能优异,与C/C复合材料具有良好的物理化学相容性,故常用作抗氧化涂层材料[9]。此外,SiC氧化后生成的SiO2在高温环境中具有流动性,对SiC涂层表面结构缺陷能起到一定的封填作用,从而有效地阻止氧气扩散进入C/C基体[10]。化学气相沉积法(CVD, chemical vapor deposition)是现今制备致密、均一SiC涂层最有效的方法,但由于CVD SiC与C/C复合材料仅以范德华力结合,故二者界面结合强度较差,C/C复合材料服役过程中SiC涂层受热应力作用容易出现开裂甚至剥落。通过改变涂层与基体材料的界面结构以形成过渡层是改善涂层与基体材料界面结合的有效途径。为了保证材料良好稳定的力学性能,实际应用中使用的C/C复合材料往往具有较高密度和较低孔隙率[11],CVD SiC涂层制备过程中,气相SiC分子扩散能力有限,难以扩散进入基体内部,这将加剧界面热膨胀系数不匹配现象,从而使CVD SiC涂层的抗氧化性能降低。为了形成较好的过渡层界面结构,SHAN等[12−13]在预氧化后的C/C复合材料表面分别制备SiC涂层和Si-Mo-Cr涂层,与不经过预氧化处理制备的涂层相比,材料的抗氧化性能大幅度提升。为进一步优化预氧化处理工艺,改善C/C复合材料的高温抗氧化性能,本文先对C/C复合材料进行预氧化处理,然后采用CVD 法制备SiC涂层,研究预氧化处理时间对CVD SiC涂层的微观结构与抗氧化性能的影响,优化预氧化处理工艺,为C/C复合材料表面SiC抗氧化涂层的制备提供实践指导。
1 实验
1.1 C/C复合材料与SiC涂层制备
首先采用化学气相渗透法将密度为0.45 g/cm3的准三维炭纤维预制体增密至1.65 g/cm3,再以酚醛树脂作为基体炭的前驱体,通过“浸渍–固化–炭化”将C/C复合材料增密至1.78 g/cm3。将制得的材料加工成尺寸为10 mm×10 mm×10 mm的抗氧化试样,用800目砂纸打磨试样表面,然后超声波清洗,烘干备用。
将C/C复合材料样品分为3组,置于温度为900 ℃的马弗炉中,分别于3,6和9 min时迅速取出,得到预氧化后的C/C复合材料。然后通过化学气相沉积法在3组预氧化后的C/C复合材料表面制备SiC涂层,以甲基三氯硅烷(MTS)为气相前驱体,氢气和氩气分别为载气和稀释气体,MTS、氢气、氩气的流量分别为100,80和360 L/h,沉积温度为1150℃,沉积时间为15 h。
1.2 性能测试
涂层试样的抗氧化性能测试在马弗炉中进行,测试温度为1 500 ℃。将试样置于刚玉坩埚内,推入恒温区,定期将试样从炉中取出,并用精度为0.1 mg的电子分析天平称量试样的质量。通过式(1)计算试样的质量损失率,并绘出氧化质量损失率随氧化时间的变化曲线。
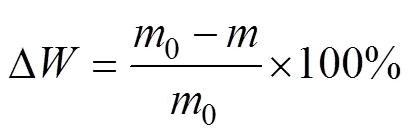
式中:Δ为试样氧化质量损失率;0为试样氧化测试前的质量;为试样氧化后的质量。
采用Quanta FEG250型扫描电镜分析C/C复合材料及SiC涂层试样的微观结构,并结合背散射(BSE)探头观察界面结构。用Jobin Yvon LabRAM Aramis型拉曼光谱仪表征基体炭与炭纤维的石墨化度。采用WYKO NT9100型光学轮廓仪测量涂层表面的粗 糙度。
2 结果与分析
2.1 C/C复合材料的微观结构
考虑到C/C复合材料为多相非均质材料,先对预氧化后C/C复合材料的不同区域微观形貌进行分析,研究C/C复合材料表面炭纤维束区域和热解炭富集区域的形貌。炭纤维束区域又分为炭纤维与材料表面垂直区域(简称纤维垂直方向)和炭纤维与材料表面平行区域(简称纤维平行方向)。
C/C复合材料中纤维束内的炭纤维之间孔隙较小,纤维束内的孔隙很容易被填充。图1为预氧化处理后C/C复合材料中纤维束垂直方向和平行方向的表面SEM形貌。由图可见,经过不同时间预氧化后,材料表面纤维束的形貌存在较大差异。图1(a)和图1(b)分别为预氧化3 min后纤维垂直方向和平行方向的表面形貌。由图1(a)可知,此时C/C复合材料刚开始氧化,氧化最先发生在炭纤维/基体炭界面上,炭纤维与基体炭间已经出现空隙,但空隙尺寸较小,由图1(b)可见纤维平行方向的材料表面依然较致密。图1(c)和图1(d)分别为预氧化6 min后纤维垂直方向和平行方向的表面微观形貌,与预氧化3 min相比,此时炭纤维与基体炭之间出现较宽较深的沟壑(图1(c)),试样表面纤维直径缩小,呈现出“圆台”状(图1(d))。预氧化9 min后(图1(e)和图1(f)),纤维之间的基体炭已经被消耗殆尽,仅留下炭纤维裸露在材料表面(图1(e)),炭纤维端头呈明显的“针尖状”(图1(f))。
图2为C/C复合材料中基体炭与炭纤维的拉曼光谱图,可以发现二者拉曼光谱的D峰和G峰均位于约1 356 cm−1和1 597 cm−1的位置。D峰是由于炭材料内部结构缺陷所引起,而G峰的产生是由于芳香碳平面内碳原子的E2g对称振动[14]。D峰越高表明炭材料内部结构缺陷越多,G峰越高表示炭材料内部原子排列越规整,用D峰与G峰强度的比值D/G表征炭材料的石墨化度,通常D/G值越大,材料石墨化度越低,结构缺陷越多,反之亦然。从图2可见,基体炭的D峰明显大于G峰,D/G=1.48,而炭纤维的G峰略大于D峰,D/G=0.92。由于研究采用的C/C复合材料未经高温石墨化处理,因此材料中基体炭的石墨化度比炭纤维低,基体炭内部结构缺陷更多,这与计算所得的D/G值相符。从图1可知,对于不同时间氧化预处理的C/C复合材料,其表面炭纤维束的微观形貌不同,可用炭纤维石墨化度高于基体炭来说明。预氧化3 min时,氧化反应最先发生在基体/纤维界面上,这是因为C/C复合材料制备过程中,制备工艺或炭纤维与基体炭热膨胀系数差异等因素,导致纤维/基体界面上存在大量缺陷,这些界面处的碳原子活化能较高,易于吸附空气中的氧分子,因此氧化反应最先发生在界面处。预氧化6 min和9 min后,氧化沿着界面向纤维与基体进行,但炭纤维的氧化速率明显小于基体炭,这是由于基体炭的石墨化度更低,内部结构缺陷更多,这些结构缺陷具有较高的能量,因而更容易受到氧分子的化学进攻,氧化速度更快。
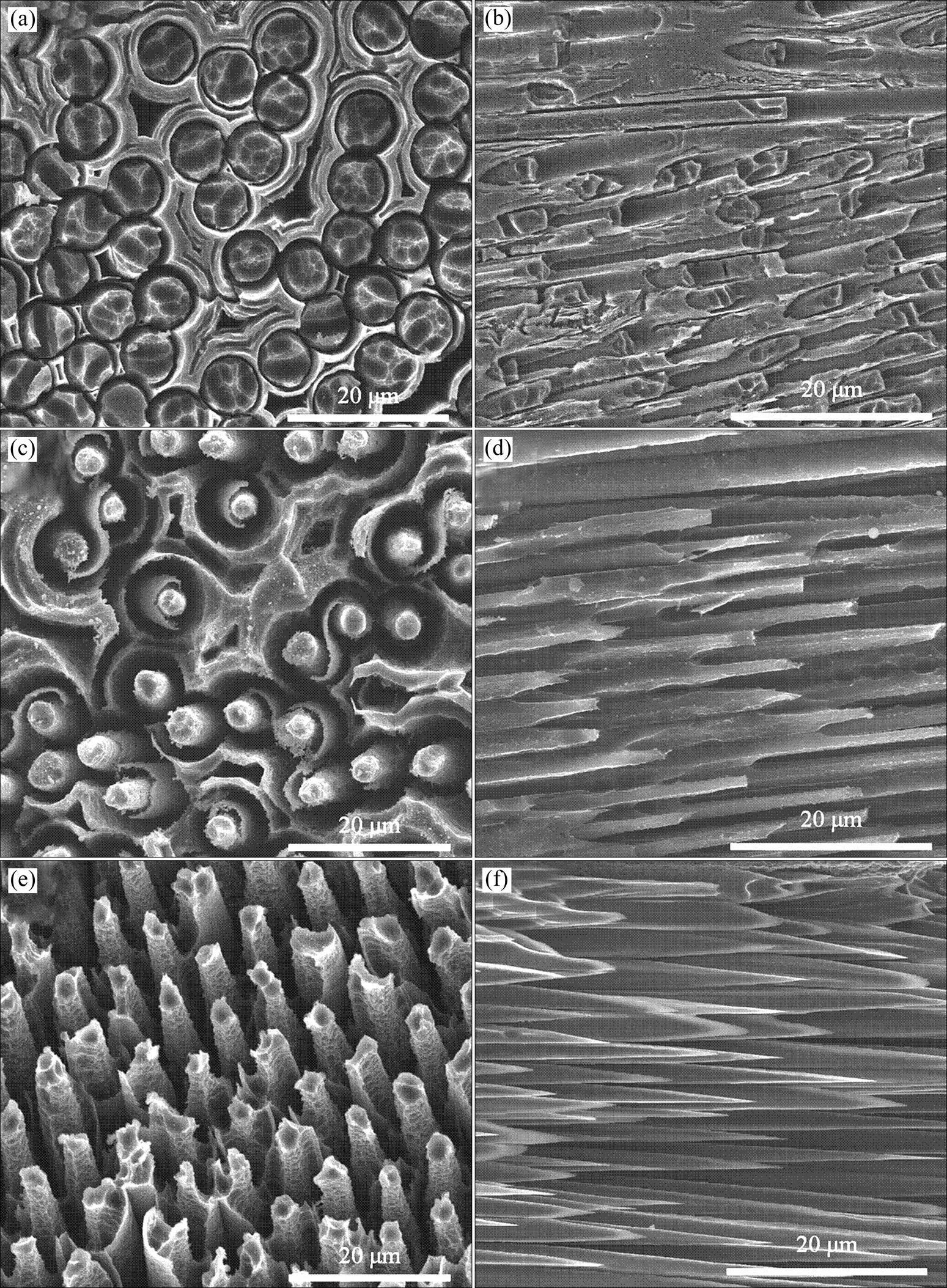
图1 预氧化不同时间后的C/C复合材料表面纤维束的SEM形貌
(a), (b) 3 min; (c), (d) 6 min; (e), (f) 9 min
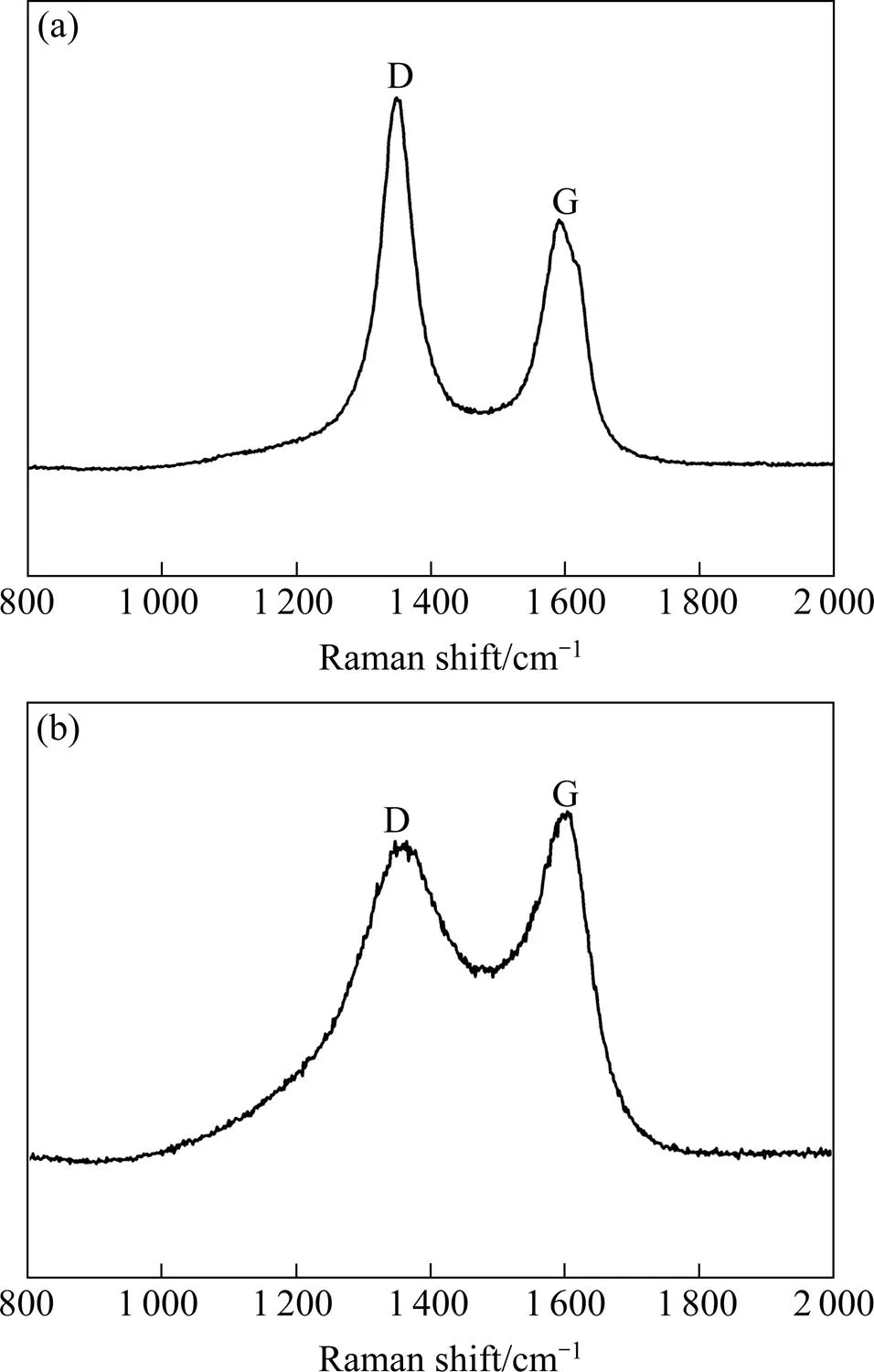
图2 C/C复合材料中基体炭与炭纤维的拉曼光谱
由于C/C复合材料制备过程中相邻炭纤维束之间存在相对较大的空隙,经碳源气体长时间渗透后,这些大空隙内往往是热解炭富集的区域。图3为热解炭富集区域预氧化不同时间后的微观形貌。由图可见,经过3 min预氧化后,热解炭已开始呈现出层片状结构,但层间距较小,结构致密。预氧化6 min和9 min后,层片之间的界限逐渐明显,热解炭结构变得疏松,说明随预氧化时间延长,越来越多的片层被氧化。

图3 预氧化不同时间后热解炭的微观SEM形貌
(a) 3 min; (b) 6 min; (c) 9 min
为了定量描述三组试样表面微观结构的变化,采用光学轮廓仪来表征预氧化后材料的表面粗糙度,并与未经预氧化处理的C/C复合材料进行对比。图4为各组试样的表面三维形貌等高图,测得的表面粗糙度a列于表1。对于抛光后未预氧化的C/C复合材料(图4(a)),其表面较平整,因此图中颜色单一、均匀,a仅为653.17 nm,蓝色部分的出现是因为试样内部不可避免地存在孔隙。预氧化3 min和6 min后,试样表面粗糙度增大,a分别达到1.18和1.48 μm。预氧化3 min后试样表面等高图中明显出现红黄相间,预氧化6 min后出现黑色区域,这是由于该处凹坑较深,白光无法探测到,导致该处呈现黑色,因此仪器检测到的表面粗糙度小于其实际值。同理,预氧化9 min后的试样表面等高图中出现更多黑色区域,虽然a仅为1.18 μm,但其实际值远大于测量值。
2.2 SiC涂层的微观结构
图5为采用化学气相沉积法在三组试样表面制备的SiC涂层的截面形貌。由图可见,涂层较致密,厚度约为80~110 μm。将预氧化时间为3,6和9 min条件下制备的SiC涂层试样分别命名为S3,S6和S9。由图5(a)可见S3的基体/涂层界面大致呈直线,涂层对基体的嵌入不明显,嵌入深度较浅,约为10 μm,这相比于涂层厚度80~110 μm,嵌入深度非常有限。与涂层试样S3相比,S6和S9涂层嵌入基体的深度大大增加,SiC扩散进基体材料的最深处均超过100μm,涂层与基体结合处呈现出犬牙交错状界面,二者具有较好的嵌入效果,这对缓解热应力、抑制裂纹扩展、增加界面结合强度十分有益。由于C/C复合材料预氧化9 min后炭纤维束内基体被消耗殆尽(图1(e)),这为涂层制备过程中气相SiC分子扩散进入基体材料提供了足够的空间,故S9试样中沉积进入C/C复合材料内部的SiC陶瓷相明显比S6中更多。
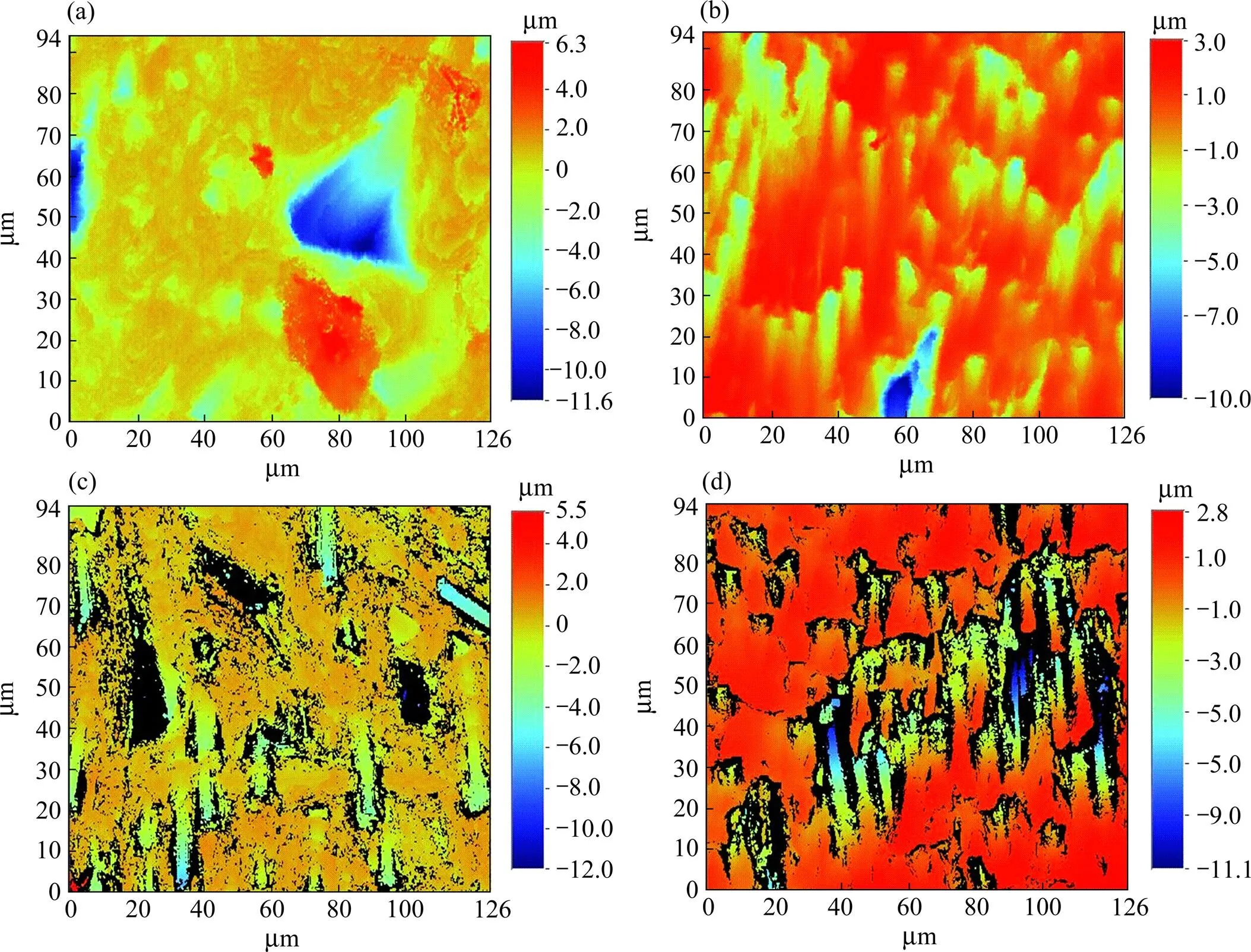
图4 C/C复合材料及其预氧化不同时间后的三维形貌等高图
(a) Without pre-oxidation; (b) 3 min; (c) 6 min; (d) 9 min

表1 C/C复合材料预氧化不同时间后的表面粗糙度
图6为涂层试样的表面SEM形貌。从图6(a)可知,涂层中SiC晶粒间结合紧密,涂层致密,未发现孔洞。将SiC涂层进行局部放大(图6(b)),该涂层由典型的“圆锥状”结构堆垛而成,“圆锥状”结构又由更小的颗粒堆积而成。此外,各“圆锥状”结构顶端均出现规律一致的放射状线条,这可能与CVD SiC的沉积机理相关。尽管CVD SiC涂层表面未观察到明显孔洞,但由于基体与涂层热膨胀系数的差异,涂层表面不可避免地出现裂纹。对比图6(c),(d)和(e)看出,制备涂层前对基体材料预氧化处理时间不同,涂层表面裂纹的宽度存在明显差异。随预氧化时间增加,涂层表面裂纹宽度变窄。这是因为预氧化时间越长,SiC陶瓷在基体材料中嵌入深度更大,有利于减小基体至涂层表面在材料组成上产生的梯度,从而缓解热应力,减小裂纹宽度。涂层表面较窄的裂纹宽度不仅能抑制氧气的扩散速率,还能在较短时间内自愈合,这对涂层更好地保护基体,提高C/C复合材料的抗氧化性能非常有利。

图5 C/C复合材料表面SiC涂层的SEM截面形貌
(a) S3; (b) S6; (c) S9

图6 C/C复合材料SiC涂层的表面SEM形貌
(a), (b) SiC grains; (c), (d), (e) Cracks in S3, S6 and S9, respectively
2.3 抗氧化性能
图7为3组涂层试样于1 500 ℃空气中恒温氧化的质量变化曲线。由图可知,S3试样的抗氧化性能最差。氧化21 h后,S6与S9的质量损失率分别为2.34%和1.89%,明显低于S3的质量损失率,且S9的抗氧化性能略强于S6。然而,从第21 h起,S9的质量损失速率明显比S6快,氧化26 h后S6与S9的质量损失率分别为4.11 %和5.39 %。
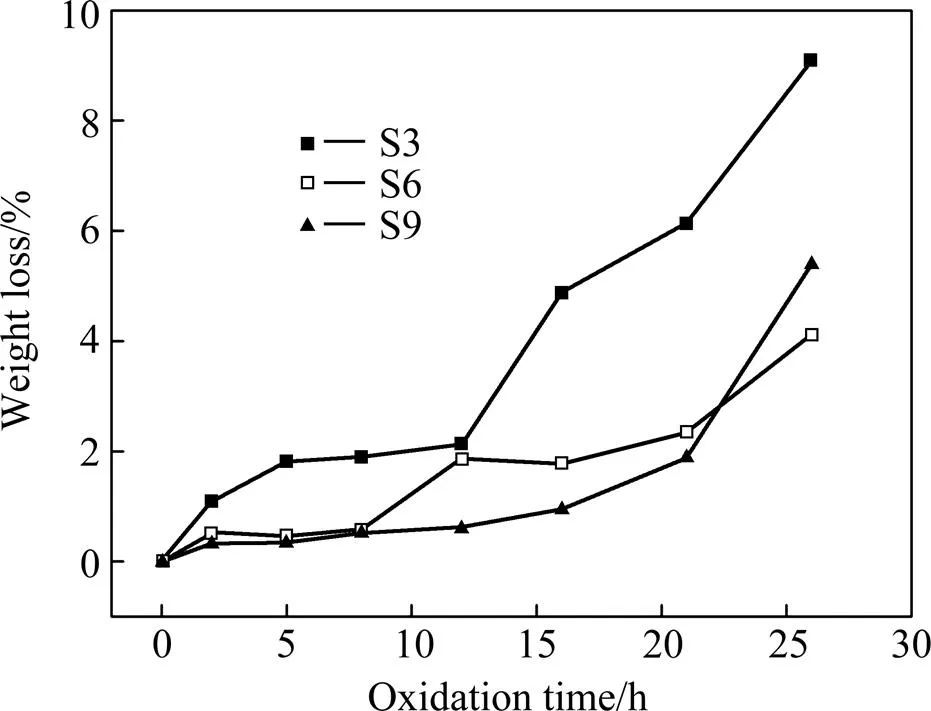
图7 SiC涂层试样的等温氧化质量损失率曲线
氧化实验前期,尽管3组试样均出现质量损失,但S3的损失速率明显比其它2组更快,且S3由初始状态至质量稳定状态经历的时间(0~5 h)比S6和S9 (0~2 h)更长,这是由于S3涂层表面的裂纹最宽,熔融态SiO2一时难以封填其表面裂纹。
当试样的质量维持相对稳定后,涂层中多数孔隙裂纹被熔融态SiO2封填,此时氧气在涂层中的扩散速率极低。然而,各组试样质量维持稳态的时长有较大差别,氧化12 h后S3开始加速氧化,质量迅速减小,而S6和S9质量维持相对稳定的时间明显更长,21 h后质量损失率才开始显著增大。图8为涂层与基体的钉扎效应示意图。S3提早失效的根本原因如下:S3的涂层对基体的嵌入深度较浅,使得界面附近存在较大的成分梯度,因此产生更大的热应力(图8中τ3>τ6,τ3>τ9。为热应力,方向水平),热应力在涂层与基体的界面处可分解为沿界面方向的剪切应力和涂层对基体的正应力,由于较大的嵌入深度可增加基体与涂层之间界面的面积,使基体与涂层的界面更曲折(图8中α3<α6,α3<α9,α为热应力方向与界面之间夹角),S3中沿界面方向的剪切应力大于S6和S9,故S3相比S6和S9更容易滋生裂纹,因此最早失效。
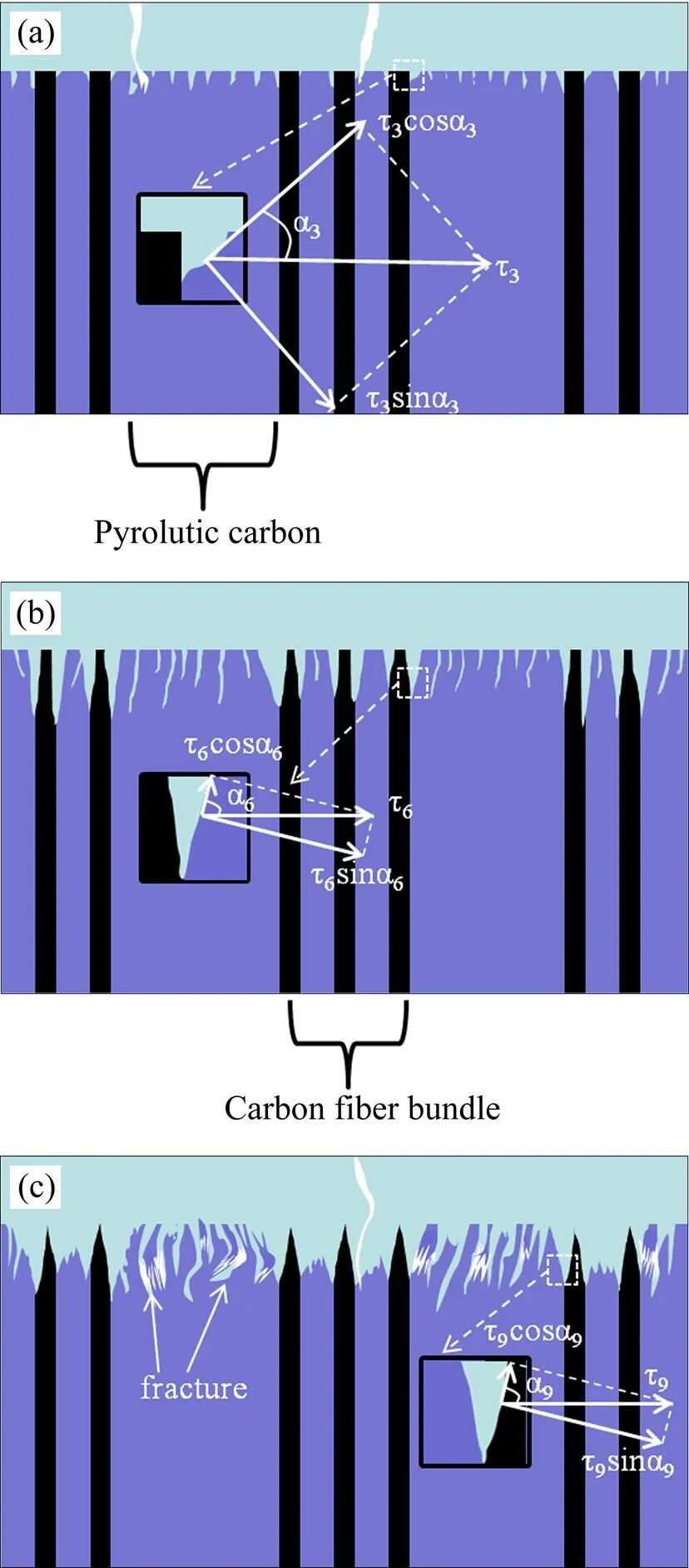
图8 涂层的钉扎效应示意图
(a) S3; (b) S6; (c) S9
图9为涂层试样在1 500 ℃环境下氧化26 h后的表面SEM形貌。由图可知,S3的表面裂纹(图9(a))远远宽于其它2组试样,这与前面所述S3中界面受较大剪切应力而易产生裂纹相符。
图10(a)为S3氧化26 h后的截面形貌图,可见涂层与基体此时已经完全脱粘,且涂层中形成了尺寸较大的贯穿性裂纹。对脱粘界面进行局部放大后(图10(d))发现,界面处原有的基体炭已全部被氧化,仅留下原本嵌入基体内部的笋尖状SiC陶瓷,说明氧气侵入基体后优先与炭材料发生反应,预氧化3 min制备的涂层对基体的保护作用有限。图10(b)、(c)分别为S6与S9氧化后的截面形貌。很明显此时S9的基体也受到了一定程度的损伤(图10(c)),涂层中形成贯穿性裂纹,但其尺寸较S3的小,涂层/基体界面附近结构疏松,热解炭被氧化后露出的炭纤维清晰可见。与S3和S9相比,S6的结构相对致密(图10(b)),但涂层/基体界面上也出现了少许微孔。

图9 涂层试样1 500℃等温氧化后的表面SEM形貌
(a) S3; (b) S6; (c) S9

图10 涂层试样在1 500 ℃等温氧化后的截面SEM形貌
(a), (d) S3; (b) S6; (c), (e) S9
由热解炭预氧化后的形貌图可知(图3),S9的基体材料表面热解炭结构疏松,而S6的基体材料中热解炭结构相对较为致密。氧化21 h以前,尽管S6与S9中涂层对基体能产生较好的“钉扎效应”,但通过分析发现,对于热解炭富集区域,虽然沉积在层片状热解炭间的SiC陶瓷能缓解热应力,但由于S9的基体材料经过预氧化后热解炭结构疏松且热解炭自身脆性较大,故该层片状热解炭力学性能较差,受较长时间热应力作用后容易断裂。图10(e)和图8(c)分别为S9的基体材料中层片状热解炭断裂后的SEM形貌与断裂示意简图,层片状热解炭发生整体断裂,进而降低涂层与基体结合强度致使涂层中出现缺陷。故与S6相比,S9氧化21 h后质量损失速率有所增加。
3 结论
1) 对C/C复合材料分别进行3,6和9 min的预氧化处理,氧化最先发生在纤维/基体界面上,材料表面呈现多孔结构,且随预氧化时间增加,基体炭氧化速率较炭纤维更快,表面粗糙度增大。
2) 分别在预氧化后的C/C复合材料表面制备CVD SiC涂层,涂层结构致密,且随预氧化时间延长,涂层对基体的嵌入深度增加,涂层表面裂纹变窄。
3) 预氧化3 min的涂层试样1 500℃空气中抗氧化性能最差。氧化21 h后,预氧化6 min与9 min的涂层试样中涂层对基体能产生较好的“钉扎效应”二者质量损失率较低。氧化21 h后,预氧化9 min涂层试样中涂层−基体界面附近层片状热解炭易发生断裂进而导致涂层产生缺陷,出现加速质量损失现象。
[1] 黄伯云, 熊翔. 高性能炭/炭航空制动材料的制备技术[M]. 长沙: 湖南科学技术出版社, 2006: 1−2. HUANG Baiyun, XIONG Xiang. Manufacturing of carbon/carbon composites for aircraft brakes[M]. Changsha: Hunan Science and Technology Publishing House, 2006: 1−2.
[2] 王馨爽, 陈招科, 熊翔, 等. C/C复合材料ZrB2-SiC基陶瓷涂层的微观结构及氧化机理[J]. 中国有色金属学报, 2017, 27(8): 1670−1678. WANG Xinshuang, CHEN Zhaoke, XIONG Xiang, et al. Microstructure and oxidation mechanism of ZrB2-SiC ceramic coating on C/C composite material[J]. The Chinese Journal of Nonferrous Metals, 2017, 27(8): 1670−1678.
[3] 杨鑫, 苏哲安, 黄启忠, 等. ZrC质量分数对C/C-ZrC复合材料力学性能的影响[J]. 中南大学学报(自然科学版), 2013, 44(2): 508−514. YANG Xin, SU Zhean. HUANG Qizhong, et al. Influence of ZrC content on mechanical properties of C/C- ZrC composites[J]. Journal of Central South University (Science and Technology), 2013, 44(2): 508−514.
[4] SHEEHAN J E, AND K W B, SULLIVAN B J. Carbon-carbon composites[J]. Annual Review of Materials Research, 2003, 24(1): 19−44.
[5] WESTWOOD M E, WEBSTER J D, DAY R J, et al. Oxidation protection for carbon fibre composites[J]. Journal of Materials Science, 1996, 31(6): 1389−1397.
[6] ZHANG Y L, HE-JUN L I, KE-ZHI L I, et al. C/SiC/Si-Mo-Cr multilayer coating for carbon/carbon composites for oxidation protection[J]. New Carbon Materials, 2012, 27(2): 105−109.
[7] 刘春轩. PIP及RMI工艺制备耐烧蚀炭/炭复合材料及其性能研究[D]. 长沙: 中南大学, 2014. LIU Chunxuan. Preparation and properties of anti-ablation C/C composites prepared by PIP and RMI[D]. Changsha: Central South University, 2014.
[8] 弭群, 曹丽云, 黄剑锋, 等. 碳/碳复合材料基体抗氧化改性研究进展[J]. 兵器材料科学与工程, 2010, 32(2): 98−103. MI Qun, CAO Liyun, HUANG Jianfeng, et al. Research progress in matrix oxidation-resistance modification of carbon/carbon composites[J]. Ordnance Material Science and Engineering, 2010, 32(2): 98−103.
[9] FRITZE H, JOJIC J, WITKE T, et al. Mullite based oxidation protection for SiC-C/C composites in air at temperatures up to 1900 K[J]. Key Engineering Materials, 1997, 132/136(16): 1629−1632.
[10] HUANG J F, ZENG X R, LI H J, et al. Influence of the preparation temperature on the phase, microstructure and anti-oxidation property of a SiC coating for C/C composites[J]. Carbon, 2004, 42(8/9): 1517−1521.
[11] ZHANG W G, H TTINGER K J. Densification of a 2D carbon fiber preform by isothermal, isobaric CVI: Kinetics and carbon microstructure[J]. Carbon, 2003, 41(12): 2325−2337.
[12] SHAN Y C, FU Q G, LI H J, et al. Improvement of the bonding strength and the oxidation resistance of SiC coating on C/C composites by pre-oxidation treatment[J]. Surface & Coatings Technology, 2014, 253: 234−240.
[13] SHAN Y, FU Q, WEN S, et al. Improvement in thermal fatigue behavior of Si-Mo-Cr coated C/C composites through modification of the C/C-coating interface[J]. Surface & Coatings Technology, 2014, 258: 114−120.
[14] 王亚峰, 张明瑜, 王丽平, 等. 各向同性热解炭低温氧化行为研究[J]. 炭素技术, 2017, 36(5): 24−28. WANG Yafeng, ZHANG Mingyu, WANG Liping, et al. Low-temperature oxidation behavior isotropic pyrocarbons[J]. Carbon Techniques, 2017, 36(5): 24−28.
[15] ZHUANG L, FU Q G, ZHANG J P, et al. Effect of pre-oxidation treatment on the bonding strength and thermal shock resistance of SiC coating for C/C-ZrC-SiC composites[J]. Ceramics International, 2015, 41(5): 6956−6964.
Effect of pre-oxidation treatment time on C/C composites and its SiC anti-oxidation coating
WANG Yujie, ZHANG Mingyu, SU Zhean, TONG Kai, HUANG Qizhong
(State Key Laboratory of Powder Metallurgy, Central South University, Changsha 410083, China)
The SiC coating with embedded interface was prepared on the surface of C/C composites by pre-oxidation treatment and chemical vapor deposition. The effects of pre-oxidation time on the microstructure of C/C composites and SiC coating were investigated, and the anti-oxidation test at 1 500℃ of the coated sample was carried out. The results show that the surface of the pre-oxidized material possesses porous structure, and the oxidation rate of matrix is greater than that of fiber. As the pre-oxidation time increases, the surface roughness of C/C composites and the embedded depth of the SiC coating increase, the coating crack narrows. In the high temperature oxidation experiments, the mass loss rates of the coated samples prepared by pre-oxidation treatment of 6 min and 9 min are 2.34% and 1.89% respectively after oxidation for 21 h. The samples prepared by 6min pre-oxidation show better oxidation resistance, and the mass loss rate is 4.11% after static oxidation for 26 h. These two samples can both produce “pinning effect” on the matrix in the first 21 h oxidation test. Nevertheless after 21 h, the thermal stress damage of the laminar pyrolytic carbon in the samples prepared by 9min pre-oxidation treatment lead to the decrease of interfacial bonding strength, which is the main reason for its rapid failure.
C/C composites; pre-oxidation treatment time; SiC coating; anti-oxidation property; pinning effect; laminar pyrolytic carbon
TB332
A
1673-0224(2018)06-591-09
湖南省战略性新兴产业科技攻关与重大科技成果转化项目(2016KG4020)
;
2018−04−28
张明瑜,副教授,博士。电话:13667385511;E-mail: csuzmy@163.com
(编辑 高海燕)
猜你喜欢
石材(2022年3期)2022-06-01
原道(2022年2期)2022-02-17
智能建筑与智慧城市(2021年11期)2021-12-08
理化检验-化学分册(2020年5期)2020-06-15
粉末冶金材料科学与工程(2019年2期)2019-05-08
材料科学与工程学报(2016年4期)2017-01-15
光学精密工程(2016年2期)2016-11-07
橡胶工业(2015年8期)2015-07-29
中国光学(2015年1期)2015-06-06
中国有色金属学报(2015年8期)2015-03-13
