
不同性别大白猪肌肉全基因组高分辨率单碱基甲基化差异分析
2018-11-30 07:14郭添福张志燕姚天雄肖石军黄路生
畜牧兽医学报 2018年11期
郭添福,张志燕,陈 冬,姚天雄,肖石军,黄路生
(江西农业大学省部共建猪遗传改良与养殖技术国家重点实验室,南昌 330045)
DNA甲基化作为一种关键的表观遗传调控机制,在调节基因表达、基因组印记、细胞分化和胚胎发育等生物过程中发挥重要作用[1]。所有组织或细胞普遍存在DNA甲基化和非甲基化机制,不同性别DNA甲基化水平是否存在差异是一个普遍关注的研究热点。El-Maarri等[2]通过比较不同性别健康人体全血基因组ALU、LINE-1 重复序列的甲基化水平差异,证实其所有CpG岛的甲基化水平男性均显著高于女性。Fuke等[3]和Shimabukuro等[4]都发现,外周血白细胞男性甲基化水平显著高于女性。此外,Xiao等[5]发现,人类肝中DNMT3a表达水平在两性之间相似,而女性的DNMT3b表达水平显著高于男性。以上结果显示,性别可能是影响DNA甲基化状态的重要因素之一,但有研究认为,不同性别的DNA甲基化水平无显著差异[6-7]。白小青等[8]研究发现,30日 龄荣昌公猪的DNA甲基化含量高于母猪,但在统计上无显著性差异。随着新一代高通量测序技术的发展,全基因组甲基化测序在动物研究中越来越受到关注,但到目前为止,还未见大白猪不同性别间全基因组高分辨率单碱基甲基化水平差异分析的报道。
本研究利用全基因组重亚硫酸盐测序法(whole genome bisulfite sequencing,WGBS)对不同性别大白猪肌肉组织的甲基化水平进行了分析,初步揭示猪性别差异的表观调控机制,为深入开展不同性别猪肌肉的分子调控研究奠定理论基础。
1 材料与方法
1.1 试验材料
本试验中大白猪来自江西樟树绿环牧业集团法系育种核心群,屠宰210 日龄健康公母猪各 2头,采集背最长肌组织,置于-80 ℃冰箱中备用。
1.2 大白猪背最长肌全基因组重亚硫酸盐测序(WGBS)
1.2.1 DNA提取及样品检测 利用标准的苯酚-氯仿法提取基因组DNA,使用分光光度计测量DNA的浓度,琼脂糖凝胶电泳分析DNA降解程度以及是否有RNA污染,Nanodrop检测DNA的纯度(OD260 nm/OD280 nm比值),用Qubit对DNA浓度进行精确定量。
1.2.2 文库构建 检测合格的基因组DNA样品,首先用Bioruptor (Diagenode, Belgium) 打断成平均大小为 200~300 bp的片段,对打断后的DNA片段进行末端修复、3′端加A碱基,并连接甲基化接头;随后进行Bisulfite处理(采用EZ DNA Methylation Gold Kit, Zymo Research);用 2% 的琼脂糖凝胶电泳选择片段;用QIAquick Gel Extraction kit(Qiagen)回收DNA片段。
1.2.3 文库质量检测 文库构建完成后,用Agilent 2100 检测文库DNA片段的完整性及插入片段大小,符合预期后,使用Q-PCR方法对文库的有效浓度进行检测,以保证文库质量。
1.2.4 上机测序 DNA文库检测合格后,把不同文库按照有效浓度及目标下机数据量的需求做pooling后进行Hiseq测序,测序策略为双末端测序。测序地点为北京诺禾致源科技股份有限公司。
1.3 大白猪背最长肌全基因组测序数据的生物信息学分析
1.3.1 参考序列比对分析 将获得的原始测序序列数据(Raw data)进行过滤,包括剔除带接头的reads,剔除未能确定碱基比例超过 10% 的reads,剔除低质量的reads(质量值没有超过20的碱基数占整个read的50%以上的reads)。得到的过滤后数据通过比对软件BSMAP[9](采用默认参数)比对到猪的参考基因组上(Sscrofa 11.1版本,下载地址:ftp://ftp.ncbi.nlm.nih.gov/genomes/Sus_scrofa/Assembled_chromosomes/seq),并计算每个样品的比对率和bisulfite(BS) 转化率等信息。
目前,微波检测领域的研究主要集中在微波介质材料的研究上.溶液物质在微波场中的行为与自身的极性有着密切的关系.极化程度可以由介电常数加以表示.溶液物质的介电常数通常与外界因素如浓度,温度和频段等存在一个确定的非线性关系[4].可以利用这种关系确定液体的成分.
1.3.2 测序深度统计 每个碱基位点比对到染色体上的读长数即为该位点的测序深度。计算CG、CHH和CHG 3种序列环境(H代表A、T和C 碱基)下C位点的测序深度。
1.3.3 不同序列环境下胞嘧啶甲基化分析 对包括甲基化位点(mC)在内共 10 bp碱基统计作图(上游4个碱基,下游5个碱基),研究不同序列环境下甲基化胞嘧啶上下游的序列特征,可为甲基化预测提供依据。
1.3.4 甲基化组DMR分析 差异甲基化区域(differentially methylated region,DMR)是在多样本间甲基化程度存在差异的一段基因组区域。DMR是一种重要的表观遗传学研究对象,可通过多种途径调控基因的表达,从而影响生物学功能。本试验采用SMART软件(2.2.1版本,采用默认参数)进行DMR分析[10]。
1.3.5 聚类分析 采取层次聚类法分析样本间DMRs的相互关系及差异趋势,以探究甲基化差异的生物学意义。
1.3.6 DMRs相关基因的GO和KEGG富集分析 利用BEDTOOLS2软件(2.25.0版本)进行基因注释,得到差异甲基化基因(differentially methylated gene,DMG)。利用DAVID数据库[11]将DMGs进行生物本体分析(GO)和代谢通路(KEGG)富集分析。对DMGs进行富集分析,可以为生物学过程相关研究提供候选基因。
2 结 果
2.1 猪背最长肌全基因组参考序列比对分析
2.1.1 猪背最长肌全基因组Reads与参考基因组比对情况统计 对 4个样品进行WGBS测序,平均每个样品产出 150.765 Gb过滤后数据。经计算,平均每个样品得到 2 374 661 943 bp的唯一匹配碱基,平均唯一匹配率为 96.67%,平均BS转化率为 99.62%,30×平均覆盖率为 73.45%,结果见表1。
表1读长与参考基因组比对情况
Table1Thecomparisonofreadstothereferencegenome

指标Index公猪_1 Boar_1公猪_2Boar_2母猪_1Sow_1母猪_2Sow_2平均Average总碱基数/bp Clean base number2 456 425 2042 456 371 7242 456 394 4932 456 380 0262 456 392 862匹配上碱基数/bp Mapped base number2 439 649 4742 440 580 7932 424 302 8792 439 262 3412 435 948 872匹配率/% Mapping rate99.3299.3698.6999.3099.17唯一匹配上碱基数/bp Unique mapped bases number2 379 110 1622 381 498 5402 359 996 4842 378 042 5862 374 661 943唯一匹配率/% Uniquely mapping rate96.8596.9596.0896.8196.67BS转化率/% BS conversion rate99.5899.6399.6299.6799.62平均深度(×) Average depth26.5631.7822.9930.7128.0130× 覆盖率/% Coverage rate73.6073.6773.1473.4073.45
2.1.2 猪背最长肌全基因组测序深度统计 根据单个碱基位点的读长数计算测序深度及其累积分布。分析显示,位于测序深度为(10×)~(30×)区段的位点最多,基本呈现正态分布趋势,见图1B;超过 80% 的全基因组位点拥有 15× 以上测序深度,见图1A,说明单位点覆盖率高,测序范围广,结果可信。

图1 大白猪背最长肌基因组测序深度分布(A)和累积分布(B)
Fig.1 Genome coverage distribution(A) and cumulative distribution(B) of sequencing depth of genome in Large White pigs LD

柱状图表示各染色体读长的平均测序深度,散点图表示每条染色体读长的覆盖率
Histogram represent the average sequencing depth of reads in different chromosomes, scatter diagram represent coverage rate of reads on each chromosome
图2 覆盖率和读长在各染色体上的分布
Fig.2 Distribution of reads and coverage rate on each chromosome
2.2 猪背最长肌全基因组甲基化水平分析
2.2.1 猪背最长肌全基因组甲基化C位点分布分析 在全基因组范围约有 5% 的胞嘧啶被甲基化(mC/C)。根据胞嘧啶(C)序列特征可以将其分为CG、CHH和CHG(H代表A、T或C 碱基)3种序列环境[12],CG环境甲基化占总甲基化的比率(mCG/mC)在3种环境中最高;甲基化主要发生在CG环境,mCHG/CHG和mCHH/CHH环境中甲基化所占比例均不超过 1%;母猪的平均mCG/CG水平要高于公猪。大白猪全基因组胞嘧啶甲基化情况见表2。
2.2.2 猪背最长肌全基因组甲基化密度图谱 不同性别不同序列环境的全基因组甲基化密度见图3,结果显示,不同个体部分染色体以及不同序列环境的甲基化密度存在差异。
2.2.3 猪背最长肌全基因组染色体甲基化水平分析 所有个体不同染色体甲基化水平的差异和趋势见图4,除母猪_1的CHG与CHH环境的甲基化分布情况外,其余个体不同环境下甲基化分布趋势基本一致。所有个体的甲基化程度(mC/C)均在12号染色体最高。
2.2.4 猪背最长肌全基因组基因功能区域甲基化水平分析 为更好地理解基因3′UTR、5′UTR、外显子、内含子和启动子(转录起始位点上游2kb的区域)等不同功能元件的甲基化水平,将每个基因的各个功能区域分别等分成20个单位长度,再对所有基因的功能元件区域相应单位长度的C位点甲基化水平求平均值后进行统计,见图5,结果显示,所有个体不同基因功能元件在3种环境的甲基化水平均相似;不同基因功能元件的甲基化水平存在差异,其中,CG环境下内含子和3′UTR的甲基化水平普遍较高,5′UTR的甲基化程度较低。
表2大白猪全基因组胞嘧啶甲基化情况
Table2Genome-widemethylationlevelofcytosineinLargeWhitepigs
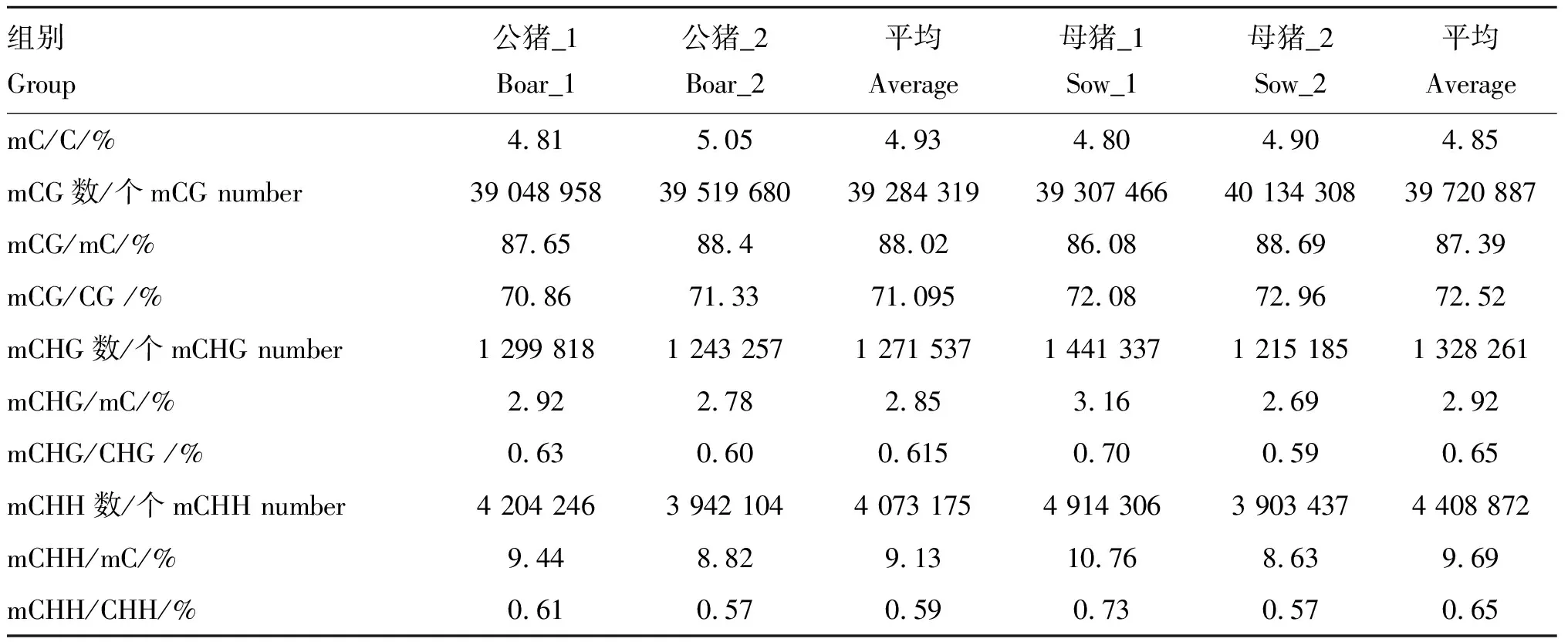
组别Group公猪_1Boar_1公猪_2Boar_2平均Average母猪_1Sow_1母猪_2Sow_2平均AveragemC/C/%4.815.054.934.804.904.85mCG数/个mCG number 39 048 95839 519 68039 284 31939 307 46640 134 30839 720 887mCG/mC/%87.6588.488.0286.0888.6987.39mCG/CG /%70.8671.3371.09572.0872.9672.52mCHG数/个mCHG number1 299 8181 243 2571 271 5371 441 3371 215 1851 328 261mCHG/mC/%2.922.782.853.162.692.92mCHG/CHG /%0.630.600.6150.700.590.65mCHH数/个mCHH number4 204 2463 942 1044 073 1754 914 3063 903 4374 408 872mCHH/mC/%9.448.829.1310.768.639.69mCHH/CHH/%0.610.570.590.730.570.65
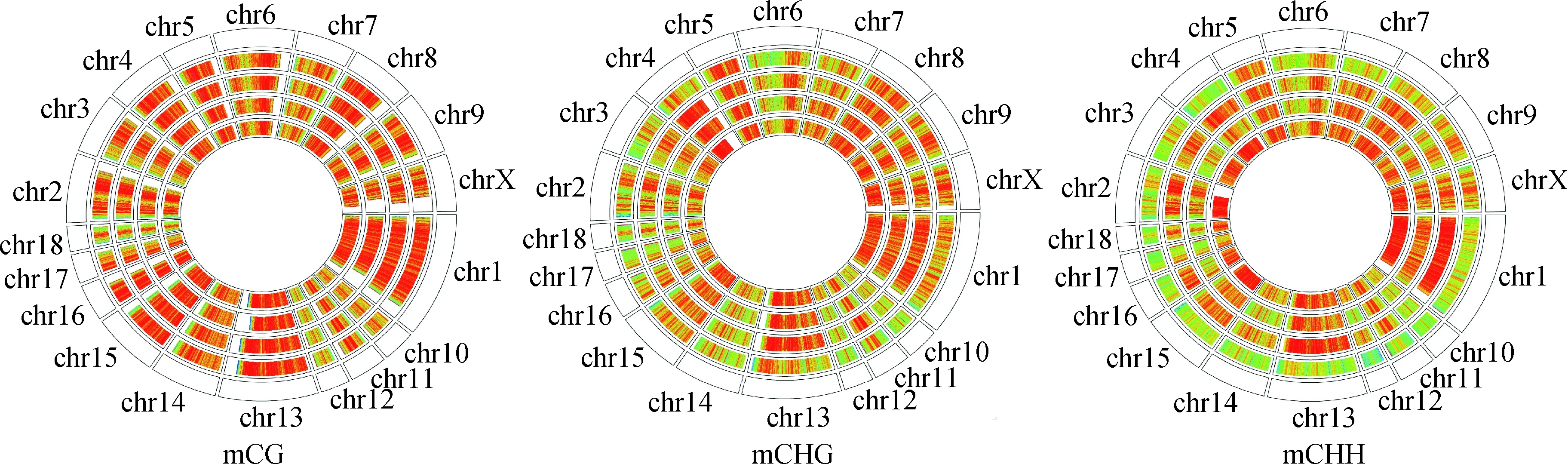
圈由外向内依次为参考基因组、公猪_1、公猪_2、母猪_1和母猪_2。红色和绿色分别代表高甲基化水平和低甲基化水平
The circles from the outside to inside represent reference genome, boar_1, boar_2, sow_1 and sow_2, respectively. Circles display high methylation level in red and low methylation level in green
图3 不同序列环境(mCG、mCHG和mCHH)甲基化密度图谱
Fig.3 Methylation density maps of mCG, mCHG and mCHH
横坐标代表染色体号,纵坐标代表不同序列环境下各染色体上甲基化的平均水平
The abscissa represent different chromosomes, and the ordinate represent methylation level of different chromosomes in different contexts
图4 C位点平均甲基化程度在染色体上的分布
Fig.4 The average level distribution of methylation at C on chromosomes
2.3 猪背最长肌全基因组在不同序列环境下胞嘧啶甲基化分析
通过对不同序列环境下甲基化位点的序列偏好分析发现,mCHG中的第2位H大部分以T居多,A 次之;mCHH第2位H以C 为主,第3位H以T 为主,见图6。
2.4 猪背最长肌全基因组DMR分析
2.4.1 猪背最长肌全基因组DMR鉴定结果及结构注释 在不同性别大白猪的背最长肌组织中鉴定出CG、CHH和CHG环境下显著的DMRs分别为118、0和1511个,共鉴定到1 629个DMRs。DMRs长度主要集中在20~300 bp,最长的出现在第 4号染色体上,达到 1 104 bp;越长的DMRs出现的频率越低,见图7A;性别差异分析显示,母猪的DMRs甲基化水平明显高于公猪,见图7B;不同染色体DMRs长度分布呈多态性,见图7C。
通过对DMRs进行注释,共鉴定出 841 个DMGs,CG和CHH环境下发生甲基化最多的元件为基因间区,其次是内含子区,具体结果见图8(图中不同颜色代表不同功能元件所占的比率)。
2.4.2 猪背最长肌全基因组DMRs聚类分析 对不同样本之间的DMRs进行聚类分析发现,相同性别的DMRs聚类在一起,不同性别间DMRs聚类差异显著,见图9。
2.4.3 猪背最长肌全基因组DMGs相关GO和KEGG富集分析 经过GO富集分析,共检测到显著相关的GO条目(P<0.05)171 个,涉及生物学过程、细胞组成以及分子功能3大类,这些DMGs显著富集在轴突导向(GO: 0007411)、细胞连接(GO: 0030054)、细胞黏附(GO: 0050839)等相关过程。图10 展示了 30 个最显著GO条目。对不同性别DMGs进行KEGG富集分析,共得到 10 个相关信号通路(P<0.05),显著富集在ECM受体互作(hsa04512)等通路,详情见图11。

将每个基因的各个功能区域分别等分成20个单位长度,再计算每个单位长度的甲基化水平求平均值绘制成折线图。横坐标代表不同的基因组功能元件,纵坐标为不同序列环境下各功能元件的甲基化水平。不同颜色代表不同序列环境(CG、CHG和CHH)
Each element of each gene was divided into 20 bins and the average of all bins in each gene elements was obtained. The abscissa represent different gene elements, and the ordinate represent methylation levels of elements in different sequence contexts. Different colors represent different sequence contexts (CG, CHG and CHH)
图5 甲基化水平在不同基因组功能元件上的分布
Fig.5 Distribution of methylation level in different genomic components
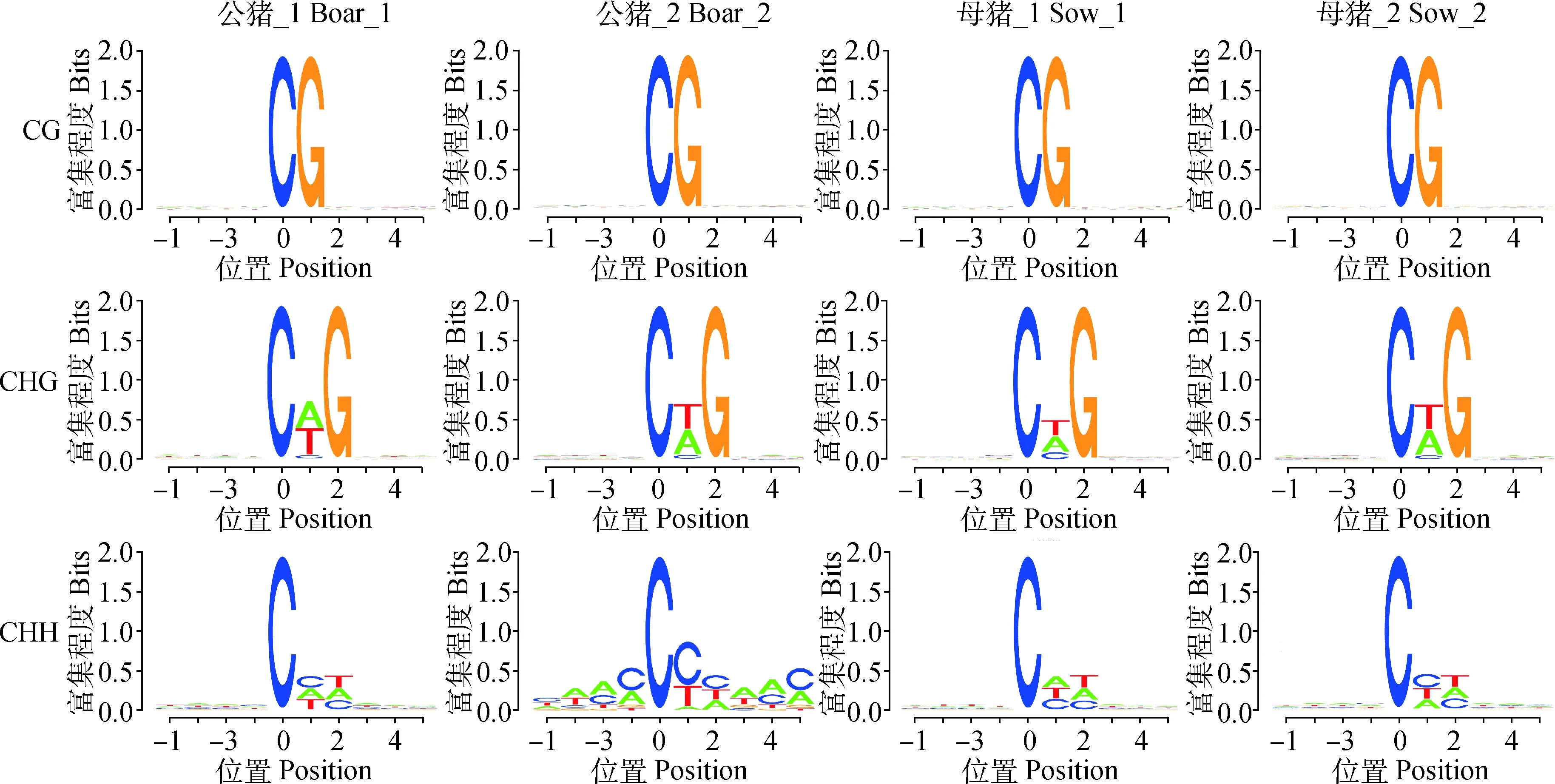
横坐标为碱基位置,mC的位置定义为0,纵坐标为相应位置碱基的富集程度,每个位置的总高度为碱基的序列含量
The abscissa represent bases position, the position of mC is defined as 0, and the ordinate represent the degree of base enrichment, the total height of each position is the sequence content of the base
图6 不同序列环境下的胞嘧啶甲基化
Fig.6 Cytosine methylation in different sequence contexts
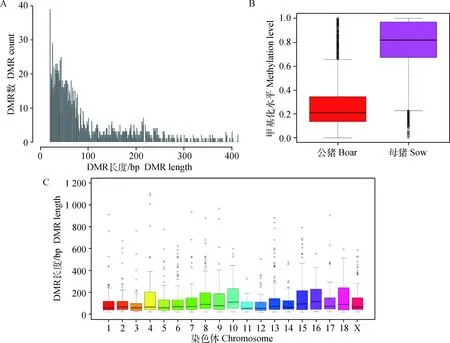
A图横坐标代表DMRs长度,纵坐标代表对应DMRs长度的频率;B图横坐标为不同性别,纵坐标代表DMRs甲基化水平分布;C图横坐标代表染色体编号,纵坐标代表DMRs长度分布
The abscissa represents DMRs length, the ordinate represents the frequency of different DMRs length in figureA;The abscissa represents gender, the ordinate represents the methylation level of DMRs in figureB;The abscissa represents different chromosome, and the ordinate represents the distribution of DMRs length in figure C
图7 DMRs长度和水平分布
Fig.7 Distribution of DMRs length and level
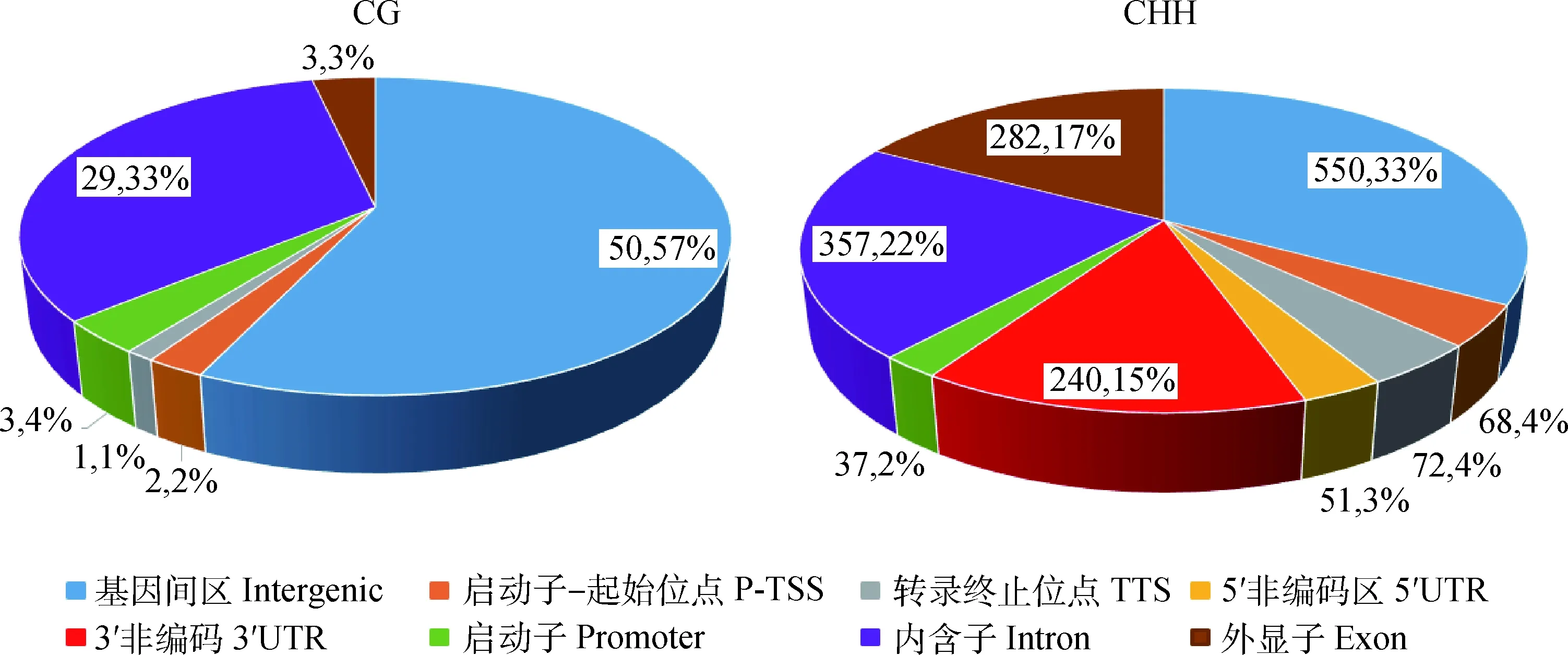
图8 不同序列环境DMRs所在基因功能元件的百分比
Fig.8 The percentage of DMRs in different gene elements in different sequence contexts
为了更好地理解DNA甲基化与不同性别肌肉差异的相关性,本研究设置2个限制因素进行候选基因筛选:1)选择GO分析中富集于肌肉相关通路的DMGs;2)第一步选择的DMGs需要显著富集在KEGG分析中的信号通路(剔除癌症相关通路)。结果得出,有 2个GO条目与肌肉相关通路有关,即平滑肌细胞迁移负调控(GO:0014912)和肌肉器官发育(GO:0007517),包含 14 个DMGs,其中有5个DMGs富集在KEGG分析中的信号通路(DMD、ITGA7、LMNA、SEMA6D和SLIT2),具体信息见表3。

行代表个体号,列代表个体的DMR
Each row represent an individual and each column represent an individual DMR
图9 大白猪不同样本DMRs聚类分析
Fig.9 DMRs cluster analysis in different samples of Large White pigs

纵坐标代表GO条目;横坐标代表-lg(P-value)
The ordinate represents the GO terms, the abscissa represents the -lg(P-value)
图10 最显著30个GO条目的柱状图
Fig.10 The histogram of top 30 GO terms

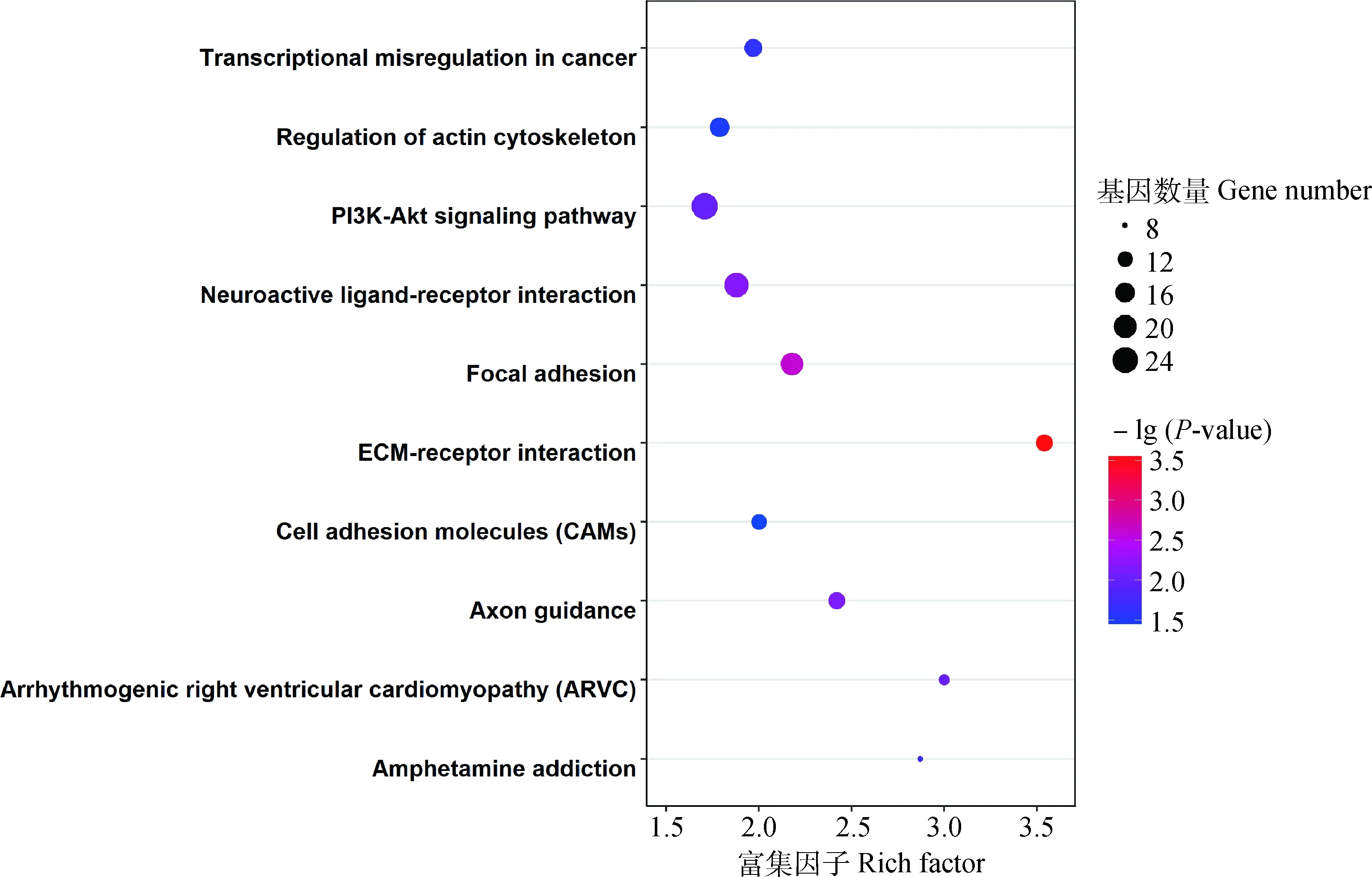
纵坐标代表KEGG条目;横坐标代表富集因子。圈的大小代表通路的基因数,圈越大,基因数越多。不同颜色代表-lg(P-value),颜色越红,-lg(P-value)越大
The ordinate represents the KEGG terms and the abscissa represents the rich factor. The size of circles represent the number of genes contained in the particular class, the larger the circle indicate the more genes. The different colors represent the -lg(P-value), the redder the circles indicate the higher -lg(P-value)
图11 KEGG条目气泡图
Fig.11 The bubble chart of KEGG terms
3 讨 论
猪是人类最主要的动物蛋白来源,肉质作为养猪生产中的一个重要经济性状,已经越来越受到研究者、生产者和广大消费者的重视。
DNA甲基化作为重要的表观遗传学现象之一,能引起染色质结构、DNA构象和转录效率等多方面的改变,从而调控基因表达[13]。近年来,猪的全基因组甲基化方面虽有研究,但主要采用的是DNA甲基化免疫共沉淀测序法[14-23]和简化代表性重亚硫酸盐测序法[23-25],虽然有极少数研究利用“金标准”重亚硫酸盐进行甲基化测序法[26-28],但覆盖度太低;主要集中在不同品种、不同组织或不同处理方式之间的甲基化差异分析,尚未见不同性别间肌肉全基因组甲基化差异的报道。本试验利用WGBS研究不同性别大白猪的背最长肌全基因组高分辨率单碱基甲基化差异,系统地比较了不同性别大白猪背最长肌全基因组甲基化的差异。
本研究发现,基因组中大约有 5% 的C位点被甲基化,不同性别间胞嘧啶甲基化水平差异不显著,与此前相关报道类似[8]。CG环境甲基化比率在3种环境中最高,与此前报道的结果一致[20-22,26-27]。不同性别功能元件区的全基因组甲基化模式相似,CG、CHG和CHH 3种序列环境的甲基化差异可能与不同遗传元件差异有关。
大部分DMRs为20~300 bp,说明小片段区域C位点甲基化对基因表达的调节起重要作用。不同序列环境下DMGs的功能元件区分布一致,CG和CHH环境主要集中在基因间区和内含子区,其他功能元件区分布均较少。出现这种现象的原因可能是,基因间区和基因的内含子区约占整个基因组的99%,所以大多数的甲基化位点出现在基因间区和内含子区域;另一方面,启动子等功能区域的甲基化会阻抑基因的表达,尤其是众多的看家基因以及早期就需要表达的基因,这些基因的功能区域都基本没有发生甲基化,而内含子和基因间区的甲基化所涉及的调控功能较少,故非功能区间的甲基化位点更多。
在GO分析中,841 个DMGs中有 757 个富集在生物学过程、细胞组成和分子功能。通过2个严格的因素(一是选择GO分析中富集于肌肉相关通路的DMGs;二是第一步选择的DMGs需要显著富集在KEGG分析中的信号通路(剔除癌症相关通路))筛选出5个可能与不同性别肌肉调节相关的DMGs:DMD、ITGA7、LMNA、SEMA6D 和SLIT2。DMD基因(抗肌肉萎缩蛋白基因)发生无义突变或框移突变而引起进行性肌营养不良症,该症是一种常见的X连锁隐性遗传的神经肌肉疾病,包括Duchenne型肌营养不良和Becker型肌营养不良,主要表现为容易跌倒,蹲站困难,丧失独立行走能力等[29-33]。ITGA7基因(整合素α7基因)是整合素家族的重要成员之一,编码的整合蛋白α7为心肌和骨骼肌层黏蛋白受体,与肌萎缩蛋白糖蛋白复合物类似,连接细胞外基质到肌动蛋白细胞骨架内部,该蛋白具有治疗DMD的潜质[34]。LMNA基因(核纤层蛋白基因)编码的核纤层蛋白对核稳定、染色质结构和基因表达有重要作用,为骨骼肌卫星细胞增殖所必需[35-38],协助维持骨骼肌的体积和强度[39],该基因突变是引起阻滞型家族性扩张型心肌病最常见的致病基因[40]。SLIT2 基因为Slit家族成员之一,编码的Slit2蛋白能阻止血小板源性生长因子诱导主动脉平滑肌细胞迁移[41]。SEMA6D 基因是Semaphorin家族成员之一,在肌肉、胚胎和脑等组织中均有表达,该基因与神经系统的发生及轴突的形成有密切关系[42];在胚胎的发育过程中,该基因通过与不同受体结合,能抑制或诱导心不同部位上皮细胞的迁移,从而在胚胎心血管的形态发生中起重要作用[43],未见该基因参与肌肉发育相关过程的报道,其生物学功能有待进一步探讨。
4 结 论
本研究全面构建了大白猪背最长肌高分辨率单碱基的全基因组甲基化图谱,并解析了性别差异的甲基化区域和基因,共检测到 1 629 个DMRs和 841 个DMGs,虽然不同性别总体甲基化水平相似,但母猪的DMRs平均甲基化水平明显高于公猪。通过富集分析,获得 171 个GO条目和 10 个KEGG条目,筛选出 5个与不同性别肌肉相关的候选基因,可为未来从遗传学和表观遗传学角度研究猪肉质性状及人类试验动物模型的开发提供重要参考。
猜你喜欢
家畜生态学报(2021年7期)2021-07-28
今日农业(2020年17期)2020-12-15
教学考试(高考生物)(2020年6期)2020-11-23
食品与生物技术学报(2020年8期)2020-01-06
学苑创造·B版(2019年5期)2019-06-14
科学24小时(2019年5期)2019-06-11
小雪花·小学生快乐作文(2017年1期)2017-03-24
中国猪业(2016年3期)2016-01-28
医学研究杂志(2015年12期)2015-06-10
中国医科大学学报(2015年10期)2015-03-01
