
咳喘宁颗粒特征图谱研究
2018-11-09 06:13符海郯刘亚丽
中国药业 2018年21期
符海郯 ,汪 慧 ,刘亚丽
(1.长江航运总医院·武汉脑科医院,湖北 武汉 430019; 2.湖北省武汉药品医疗器械检验所,湖北 武汉 430075)
中药成方制剂药味复杂,仅凭单一成分测定不能全面、综合地评价其质量,且受方剂的整体物质群难以控制等因素的制约[1-2],其质量评价是研究难点[3]。相比传统的药材鉴别技术,中药特征图谱具有能运用现代分析仪器进行分析、准确度高、重复性良好等优势,可有效控制中药质量[4-5],广泛用于中药材及其制剂的质量控制,对中药质量标准的提高及现代化的构建起着重要作用[6-7]。咳喘宁颗粒由麻黄、紫菀 、百部、甘草、苦杏仁5味中药组方,具有止咳化痰之功效,可用于伤风感冒,急、慢性支气管炎等病症。已知的咳喘宁颗粒质量研究多为单味药材、单个成分的薄层鉴别分析[8],尚无关于咳喘宁颗粒特征图谱研究的报道。本研究中根据咳喘宁颗粒处方药味的组方、成分特性,以期建立一个科学、可行的特征图谱方法,整体评价不同企业的原药材、投料及制剂的质量状况。现报道如下。
1 仪器与试药
UltiMate 3000分析型高效液相色谱仪(戴安公司),包括 LPG-3400SD四元泵,WPS-3000SL ANALYTICAL自动进样器,TCC-3000SD柱温箱,DAD-3000二极管阵列检测器;XS105DU型电子天平(十万分之一,Mettler Toledo公司);CLXXXUVM2型超纯水系统(ELGA公司);SK250LHC型超声波清洗器(上海科导超声仪器有限公司);2012版中药色谱指纹图谱相似度评价软件系统。
麻黄、苦杏仁、紫菀、百部、甘草对照药材,盐酸麻黄碱、盐酸伪麻黄碱、苦杏仁苷、甘草苷、甘草酸铵对照品,均由中国食品药品检定研究院提供,批号分别为121051-200801, 121554-200607, 120956-201107,121221-201009, 120904-201319, 171241-201101,171237-201211, 110820-201512, 111610-201006,110731-201306;咳喘宁颗粒由2个厂家提供,共22批,详见表1;乙腈(色谱纯,Merck公司),水为超纯水,其他试剂均为分析纯。

表1 咳喘宁颗粒样品信息
2 方法与结果
2.1 色谱条件
色谱柱:Tech Mate C18-ST柱(250 mm ×4.6 mm,5 μm);流动相:乙腈(A) -0.1% 磷酸(B),梯度洗脱程序见表 2;检测波长:207 nm(0~40 min),237 nm(40~55 min);流速:0.9 mL /min;柱温:30 ℃。
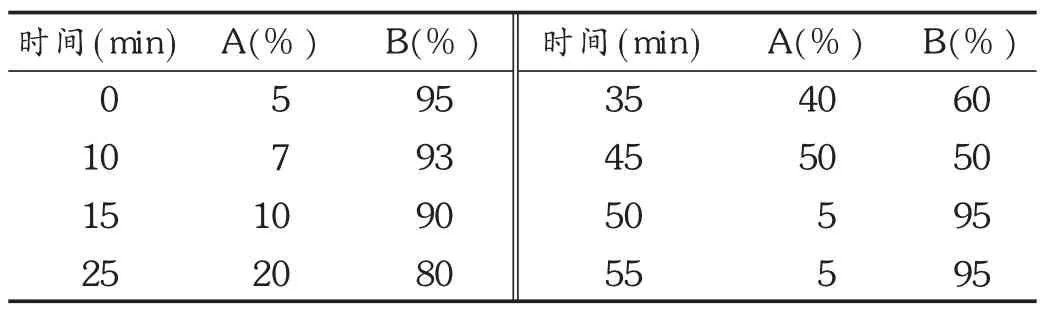
表2 流动相梯度洗脱程序
2.2 溶液制备
称取装量差异项下的样品适量,置研钵,研匀,取粉末5 g,精密称定,置标示量为50 mL的容量瓶中,加甲醇 40 mL,超声处理(功率为 100 W,频率为 50 kHz)30 min,放冷,用甲醇定容至刻度,摇匀,滤过,取续滤液,即得供试品溶液。
分别取麻黄、苦杏仁、紫菀、百部、甘草对照药材适量,按照处方比例及制备工艺制成药材提取物,按供试品溶液制备方法配制成对照药材溶液。
分别取盐酸麻黄碱、盐酸伪麻黄碱、苦杏仁苷、甘草苷和甘草酸铵对照品适量,精密称定,加甲醇溶解,得盐酸麻黄碱、盐酸伪麻黄碱、甘草苷和甘草酸铵的质量浓度均为 10 μg /mL、苦杏仁苷的质量浓度为 30 μg /mL的对照品溶液。
按照处方比例及制备工艺分别制备不含麻黄、苦杏仁、紫菀、百部、甘草的阴性样品,按供试品溶液制备方法配制阴性对照品溶液。
2.3 方法学考察
精密度试验:取样品(批号为150502),依法制备供试品溶液,按拟订色谱条件测定,连续进样6次,记录特征谱图,以1号峰(盐酸麻黄碱)为S1峰,计算1号~2号峰的相对保留时间;以6号峰(甘草苷)为S2峰,计算3号~9号峰的相对保留时间。结果各共有色谱峰相对保留时间的 RSD均小于2%(n=6),表明仪器精密度良好。
重复性试验:取样品(批号为150502),依法平行制备供试品溶液5份,按拟订色谱条件各进样1次,记录特征谱图。结果各共有峰相对保留时间的 RSD均小于2%(n=5),表明方法重复性良好。
稳定性试验:取样品(批号为150502),依法制备供试品溶液,按拟订色谱条件测定,分别于 0,2,4,8,12,24 h时进样,记录特征图谱。结果各共有峰相对保留时间的 RSD均小于2%(n=6),表明供试品溶液在24 h内基本稳定。
2.4 特征图谱的建立及其共有特征峰的归属
共有峰确定:分别精密吸取对照药材溶液、供试品溶液及阴性对照品溶液各10 μL,注入高效液相色谱仪,按拟订色谱条件测定。结果供试品溶液色谱中,共有9个特征峰,分别表征处方中的麻黄、苦杏仁、百部和甘草,处方中的紫菀未找到相应的色谱峰进行表征,确认了1号、2号峰来源于麻黄,3号、5号峰来源于百部,4号峰来源于苦杏仁,6~9号峰来源于甘草。色谱图见图1。
对照品对照法:照拟订色谱条件,对供试品溶液和对照品溶液进样分析,并对9个共有峰进行DAD扫描。通过采用对照品色谱峰结合DAD扫描图谱的方式,确定了1号峰为盐酸麻黄碱,2号峰为盐酸伪麻黄碱,4号峰为苦杏仁苷,7号峰为甘草苷,9号峰为甘草酸铵。详见图2。
特征图谱共有模式的建立及分析:通过对22批供试品特征图谱的分析,建立咳喘宁颗粒特征图谱共有模式,以盐酸麻黄碱为参比峰1,计算峰1~峰2的相对保留时间;以甘草苷为参比峰2,计算峰3~峰9的相对保留时间。结果22批样品中特征峰的相对保留时间均在规定值 ±5% 内:1.000 min(峰 1,盐酸麻黄碱,S1),1.046 min(峰 2),0.550 min(峰 3),0.740 min(峰 4),0.855 min(峰 5),0.990 min(峰 6),1.000 min(峰 7,甘草苷,S2),1.200 min(峰 8),1.374 min(峰 9)。详见图3、表3和表4。
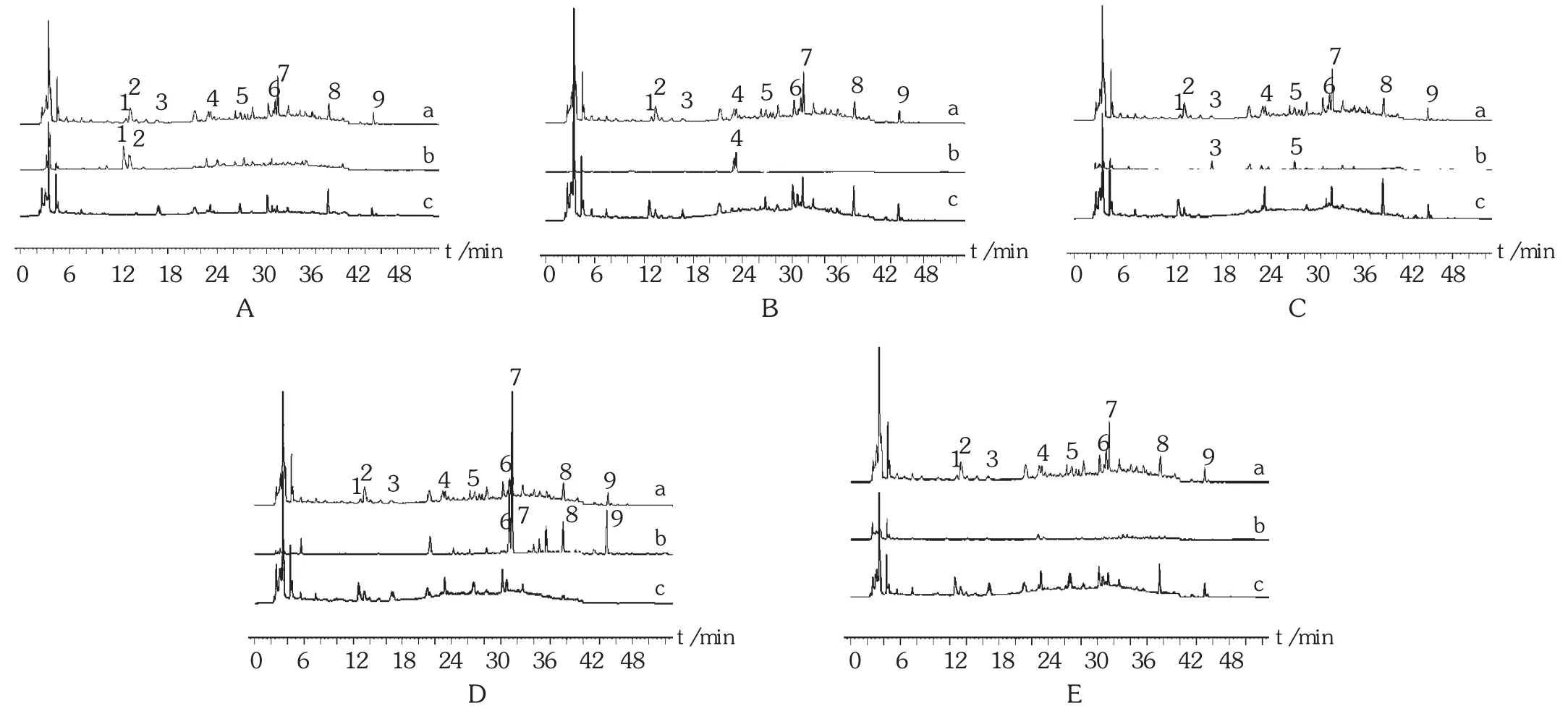
图1 特征图谱
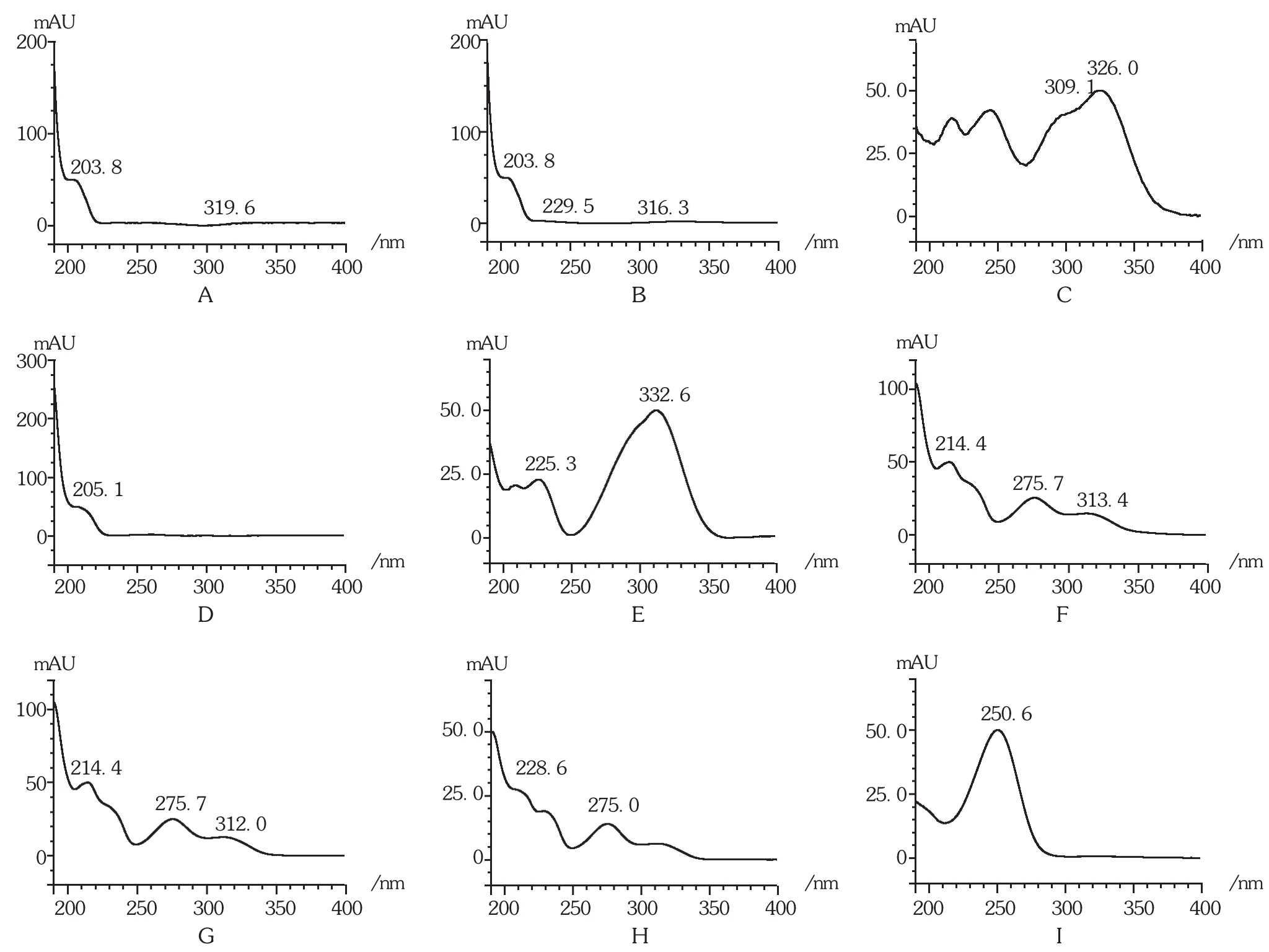
图2 特征峰DAD扫描图
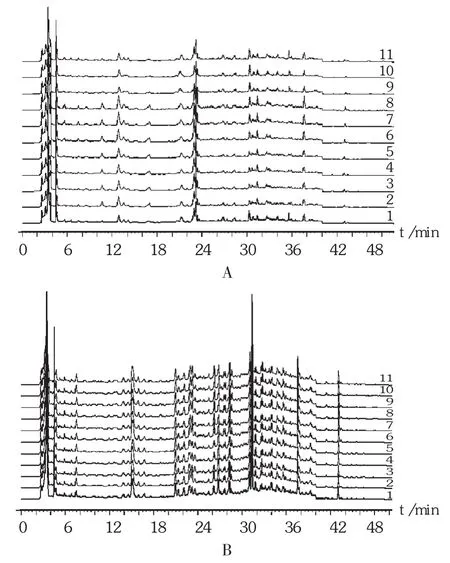
图3 22批咳喘宁颗粒特征图谱共有模式
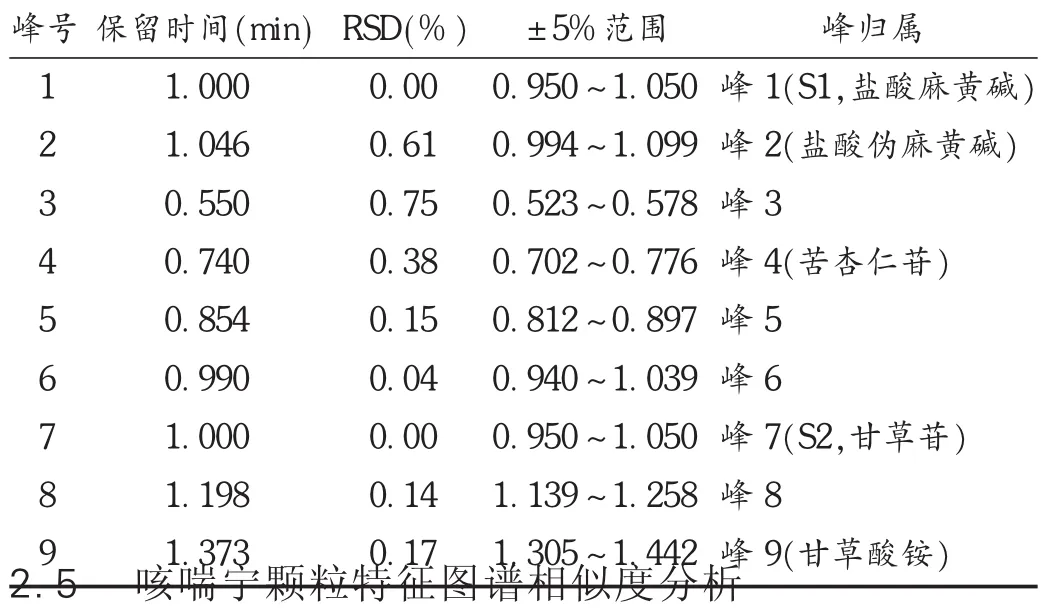
表3 22批供试品中共有峰相对保留时间及峰归属
2.5 咳喘宁颗粒特征图谱相似度分析

表4 22批供试品相对保留时间(min)
分别将2个厂家的22批样品导入2012版“中药色谱指纹图谱相似度评价软件系统”进行相似度分析。结果表明,A厂家的11批样品与该企业样品生成的对照图谱的相似度为0.933~0.999,B厂家样品与对照图谱的相似度为0.986~0.998,均大于0.90,说明2个生产厂家的产品制备工艺均较稳定,各样品一致性较好。详见表5。
3 讨论
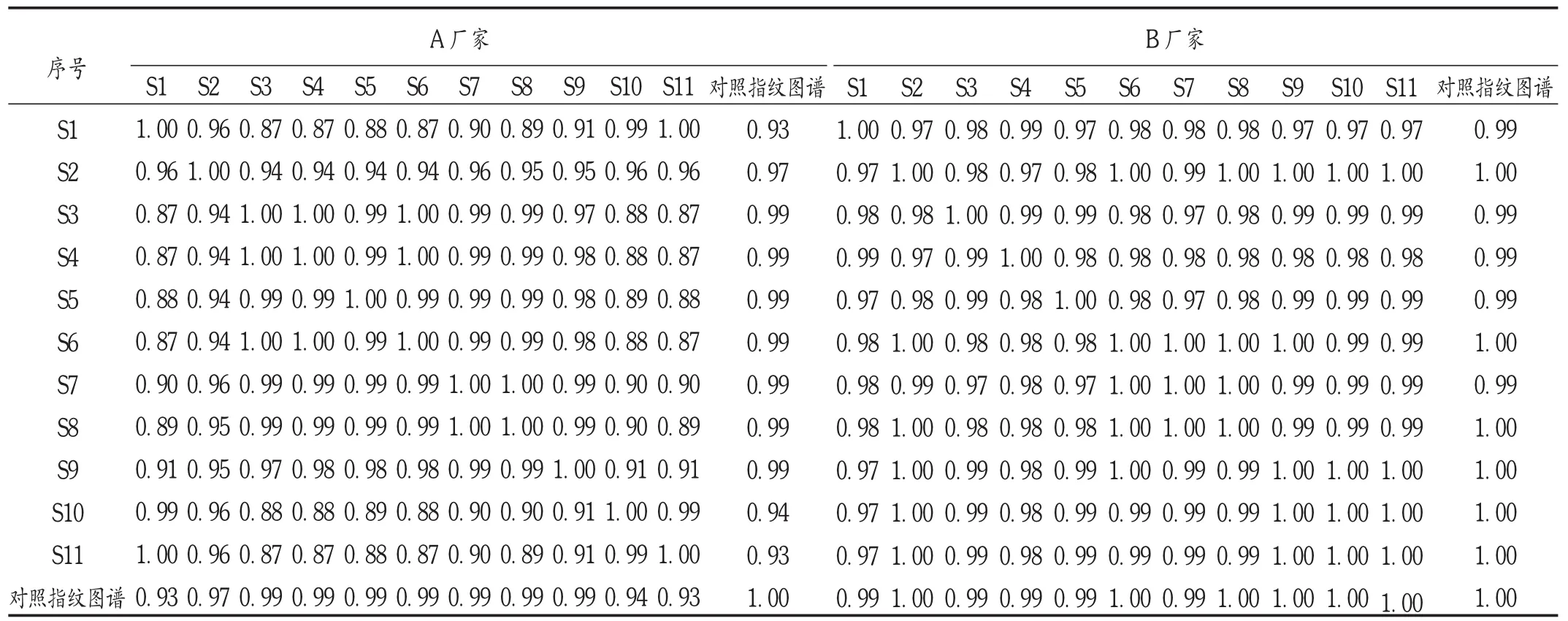
表5 A厂家和B厂家11批咳喘宁颗粒相似度分析结果
3.1 检测波长选择
咳喘宁颗粒中化学成分复杂,其紫外吸收波长各有所不同,2015年版《中国药典(一部)》[9]中麻黄、苦杏仁和紫菀含量检测波长分别为210,207,200 nm,甘草为237 nm。为了兼顾各类成分的同时测定,选用207 nm和237 nm进行双波长切换检测,考察了2种切换方式,方式 1为 0~30 min 207 nm,30~55 min 237 nm;方式 2为 0~40 min 207 nm,40~55 min 237 nm。结果表明,切换方式2所得到的色谱峰信息较丰富,故选用双波长切换方式2作为试验方法。
3.2 样品提取方法选择[10]
分别考察4种提取方法:70%甲醇超声30 min,50 mL热水溶解,纯甲醇超声30 min;甲醇-1%磷酸(70∶30)超声30 min。结果表明,各提取方法得到的液相色谱图差异不大,纯甲醇超声30 min得到的供试品溶液色谱图基线最平整,且提取方法简便,故选用其作为样品提取方法。
3.3 流动相选择
考察了甲醇-0.1%磷酸(梯度洗脱)[11]、乙腈-0.1%磷酸(梯度洗脱)2种流动相体系,发现甘草在甲醇体系中色谱峰分离效果不好,且基线不够平整,含有的特征图谱信息量较少;但乙腈体系中各药材色谱峰分离相对较好,且所含有的特征峰信息量较丰富。故选用乙腈-0.1%磷酸(梯度洗脱)作为流动相。
3.4 参照峰选择
试验发现,咳喘宁颗粒中盐酸麻黄碱和盐酸伪麻黄碱容易受pH、室温和柱温改变的影响,其保留时间会产生漂移。因此,采用了2个参照物成分计算相对保留时间。即以盐酸麻黄碱峰为参比峰1,计算峰1~峰2的相对保留时间;以甘草苷峰为参比峰2,计算峰3~峰9的相对保留时间。结果表明,各特征峰的相对保留时间基本保持稳定。
3.5 不同生产厂家咳喘宁颗粒的特征峰分析
通过对咳喘宁颗粒特征图谱共有模式进行分析,发现不同厂家生产的咳喘宁颗粒间化学成分存在明显差异:1)22批样品按特征图谱方法检测,均检出9个特征峰,符合规定,但A厂家样品的图谱整体较B厂家样品的图谱峰个数偏少,某些峰面积偏低,表现在百部的2个特征峰和甘草的4个特征峰。造成这一差异的原因,可能与各厂家投料的药材质量差异有关。2)A厂家样品色谱图中盐酸麻黄碱(峰1)的峰面积大于盐酸伪麻黄碱(峰2)的峰面积,而B厂家的样品则相反。这与两家企业所投麻黄的品种来源有关,通过文献[12]调研与特征图谱结果发现,A厂家样品所投的为草麻黄,B厂家样品所投的为中麻黄,与企业调研了解的情况一致,二者均为麻黄的法定来源。
3.6 结语
本研究中通过建立咳喘宁颗粒特征图谱,确定了9个具有鉴别意义的特征峰,指认了盐酸麻黄碱、盐酸伪麻黄碱、苦杏仁苷、甘草苷及甘草酸铵5个共有峰;通过双波长切换检测技术方法解决了咳喘宁颗粒中有效成分群紫外波长吸收差异大的问题。本方法灵敏度高、重复性好、可靠性强,可为咳喘宁颗粒质量控制和评价提供科学、有效的依据。
猜你喜欢
中国酿造(2022年2期)2022-03-10
药品评价(2021年13期)2021-09-07
中老年保健(2021年9期)2021-08-24
中成药(2020年11期)2020-12-13
Digital Chinese Medicine(2020年3期)2020-11-03
Digital Chinese Medicine(2020年2期)2020-09-26
中成药(2020年3期)2020-03-27
新传奇(2019年19期)2019-10-08
新传奇(2019年16期)2019-10-08
新传奇(2019年17期)2019-08-04
