
Genome-wide detection of selective signatures in a Duroc pig population
2018-11-06 08:19:14DlAOShuqiLUOYuanyuMAYunlongDENGXiHEYingtingGAONingZHANGHaoLlJiaqiCHENZanmouZHANGZhe
Journal of Integrative Agriculture 2018年11期
DlAO Shu-qi, LUO Yuan-yu , MA Yun-long, DENG Xi HE Ying-ting GAO Ning ZHANG Hao Ll Jia-qi CHEN Zan-mou ZHANG Zhe
1 Guangdong Provincial Key Lab of Agro-Animal Genomics and Molecular Breeding/National Engineering Research Centre for Breeding Swine Industry/College of Animal Science, South China Agricultural University, Guangzhou 510642, P.R.China
2 Liupanshui Academy of Agricultural Sciences, Liupanshui 553001, P.R.China
3 Key Laboratory of Agricultural Animal Genetics, Breeding, and Reproduction, Ministry of Education/College of Animal Sciences and Technology, Huazhong Agricultural University, Wuhan 430070, P.R.China
Abstract The Duroc pig has high adaptability and feeding efficiency, making it one of the most popular pig breeds worldwide. Over long periods of natural and artificial selection, genetic footprints, i.e., selective signatures, were left in the genome. In this study, a Duroc pig population (n=715) was genotyped with the Porcine SNP60K Bead Chip and the GeneSeek Genomic Profiler (GGP) Porcine Chip. The relative extended haplotype homozygosity (REHH) method was used for selective signature detection in a subset of the population (n=368), selected to represent a balanced family structure. In total, 154 significant core regions were detected as selective signatures (P<0.01), some of which overlap with previously reported quantitative trait loci associated with several economically important traits, including average daily gain and backfat thickness. Genome annotation for these significant core regions revealed a variety of interesting candidate genes including GATA3, TAF3,ATP5C1, and FGF1. These genes were functionally related to anterior/posterior pattern specification, phosphatidylinositol 3-kinase signaling, embryonic skeletal system morphogenesis, and oxidation-reduction processes. This research provides knowledge for the study of selection mechanisms and breeding practices in Duroc and other pigs.
Keywords: Duroc, selective signature, candidate genes, REHH
1. lntroduction
Pigs have been domesticated and artificially selected over approximately 10 thousand years to provide animal-based protein for human consumption (Groenen et al. 2012).Duroc, one of the most popular worldwide commercial swine breeds, has been subjected to strong artificial selection for their productivity, reproduction, and product quality. Genes play an important role during the process of domestication and evolution. Revealing the underlying selection mechanisms would not only benefit future pig breeding, but also facilitate the identification of porcine genes related to biological processes and traits of interest.
Rapid developments in the field of high-throughput sequencing and genotyping makes it possible to explore genomic evidence of selection, and detect candidate genes associated with economically important traits. Current approaches to detect selective signatures include the methods based on: (1) single point frequencies of select mutations such as Fay and Wu’s H-test (Fay and Wu 2000)and Tajima’s D test (Tajima 1989), (2) linkage disequilibrium(LD) such as the integrated haplotype score (iHS) (Voight et al. 2006) and the extended haplotype homozygosity (EHH)test (Pardi et al. 2002), and (3) population differentiation such as the FSTtest (Weir and Cockerham 1984). Among these methods, EHH can effectively detect positive selective signatures in a single population (Walsh et al. 2006; Zhang et al. 2006) and putative core regions by characteristics of haplotypes without ancestor allele genotypes (Qanbari et al.2010). Core regions are regions of interest in genomes can be genotyped to identify haplotypes with high EHH and high population frequencies (Sabeti et al. 2002). Furthermore,the relative extended haplotype homozygosity (REHH) test overcomes limitations of the heterogeneous recombination among chromosomal regions, which may potentially cause some false positives in EHH detection (Qanbari et al.2010). Many recent studies focused on genome-wide selective signature detection were conducted in various pig breeds, including Chinese indigenous breeds and western commercial breeds (Ai et al. 2013, 2014; Wilkinson et al.2013; Li M Z et al. 2014; Li X et al. 2014; Ma et al. 2014,2015; Wang et al. 2014; Yang et al. 2014; Moon et al.2015). Wilkinson et al. (2013) reported several genes associated with reproduction, growth, and fat deposition traits in European breeds using FSTtests. Li M Z et al.(2014) reported a number of strong signatures of selection related to disease resistance, pork yield, fertility, tameness,and body length through Tajima’s D test. Using REHH test, Wang et al. (2014) found many candidate genes in biological categories associated with brain development,metabolism, growth, and olfaction in Yorkshire and Landrace pigs.
However, only a few studies have focused on the Duroc pig (Ai et al. 2013; Wilkinson et al. 2013; Bosse et al.2014; Choi et al. 2015). These studies reported that the quantitative trait locus (QTL) harboring the ELOVL3 gene was identified by selective signature detection (Westerberg et al. 2006), which is involved in fatty acid biosynthesis(Sanchez et al. 2007; Uemoto et al. 2012), and overlaps with previously reported QTL on Sus scrofa chromosome 14(SSC14) in the Duroc pig. In addition, the Class III myosin B(MYO3B) gene was found on SSC15 by selective detection(Wilkinson et al. 2013). The aim of this study was to detect specific signatures of recent selection in the Duroc pig genome. In this study, the REHH test was implemented to scan the whole genome for detecting selective signatures in Duroc with the Illumina Porcine SNP60K Bead Chip (Ramos et al. 2009) and the GeneSeek Genomic Profiler (GGP)Porcine Chip. Our findings identify important candidate functional genes that underwent positive selection in Duroc pig.
2. Materials and methods
2.1. Population and genotypes
The Duroc pig population used in this study was maintained as a breeding herd in a farm located in the Fujian Province of China. Ear tissue of 368 Duroc pigs (348 females and 20 males) selected from 715 unrelated pigs (no common ancestor within three generations) was collected for DNA extraction. The selection criteria were as follows: (1) divide all individuals into different families based on paternity; (2)remove individuals with inconsistent pedigree records; (3)retain at least one full-sib from each family; and (4) exclude individuals from each family with more than three half-sibs.Genomic DNA was extracted using the MiniBEST Universal Genomic DNA Extraction Kit (Ver. 4.0, TaKaRa, USA)following a routine phenol/chloroform protocol. In order to identify whether genomic DNA was contaminated by RNA or proteins, the OD260/280ratio and DNA concentration of these samples were quantified with a NanoDrop 2000(ThermoFisher Scientific, USA). Samples with an OD260/280ratio between 1.7 and 2.0 and a concentration of at least 50 ng μL-1were used for genotyping.
The Illumina Porcine SNP60K Bead Chip and the GGP Porcine Chip, which contain 61 565 single nucleotide polymorphisms (SNPs; Ramos et al. 2009) and 50 697 SNPs respectively, were used for whole genome genotyping.Beagle (Ver. 3.3.1) Software (Browning and Browning 2007)was used to impute missing genotypes and infer haplotypes.SNPs with unknown positions or those located on sex chromosomes were removed from the dataset. Quality control on genotypes was performed with PLINK Software(Ver. 1.07) (Purcell et al. 2007). The criteria for quality control were: minor allele frequency (MAF)>0.01, call rate for SNP>0.90, call rate for individuals>0.90, and P-values of Hardy-Weinberg equilibrium>0.000001.
2.2. Selective signature detection
EHH is defined as the probability that two randomly chosen haplotypes carrying the candidate core haplotype are homozygous for the entire interval spanning the core region for a given locus (Sabeti et al. 2002). The EHH of a tested core haplotype t is:
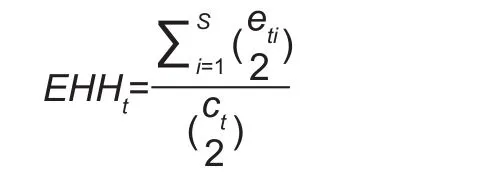
Where, ctis the number of samples for a particular core haplotype t, etiis the number of samples of a particular extended haplotype i, and s is the number of unique extended haplotypes.
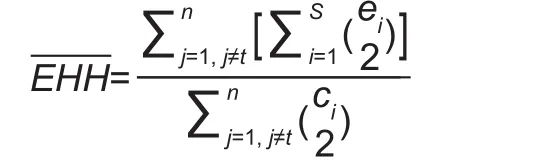
Where, n is the number of different core haplotypes.
In this study, the REHH test (Sabeti et al. 2002) was used to detect selective signatures. The REHH test, which is an extended version of the EHH test, was proposed by Sabeti et al. (2002). The REHH score is calculated as:
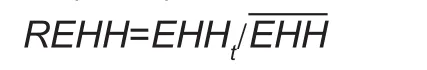
Where, the marker H value is the degree to which each added marker at a further distance causes the extended haplotype to decay for all core haplotype, and can be calculated as ‘all EHH’. Previous studies have shown that the LD extent of commercial pigs is much higher than many Chinese pig breeds (Nsengimana et al. 2004; Amaral et al.2008; Ai et al. 2013). In our study, the marker H value was assigned as 0.1 following Li X et al. (2014). The REHH values were calculated using Sweep Software (Ver. 1.0)(Sabeti et al. 2002). A core haplotype with a frequency>0.25 and REHH P-value<0.01 was treated as a significant core region.
2.3. Genome annotation
For each detected selective signature, a target region was defined by extending 250 kb both upstream and downstream of the region. Genes located within this target region were treated as candidate genes. Genome positions of candidate genes were identified based on the annotation of Sus scrofa 10.2 (https://www.animalgenome.org/blast/). RNAs and unconfirmed genes were excluded from annotated candidate genes. To further explore the biological functions of the detected selection regions, trait specific QTL were retrieved from the animal QTL database (Hu et al. 2016)for each target region.
2.4. GO terms and KEGG pathway enrichment analysis
The Database for Annotation, Visualization, and Integrated Discovery (DAVID) Version 6.8 (https://david.ncifcrf.gov/)(Huang et al. 2009; Wei et al. 2009) was used for Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway(Kanehisa and Goto 1999) and Gene Ontology (GO)(Ashburner et al. 2000) enrichment analyses. The GO terms and KEGG pathways with P-value<0.05 were considered significant.
3. Results
3.1. Marker and core haplotype statistics
A total of 65 742 SNPs were subjected to quality control. Of these, 646 SNPs were excluded due to HWE test P-values less than 0.000001. A total of 17 581 SNPs with MAF below 0.01 were filtered out. Finally, 47 515 SNPs were retained and were divided into 3 693 core regions. Descriptive statistics of the genome-wide markers are listed in Table 1.
3.2. Genome-wide REHH tests
Altogether, 3 693 core regions with total length of 523.57 Mb covering 21.36% (523.57/2 450.72) of the swine genome were defined. Among these core regions, the highest distribution was observed on SSC1, SSC9, and SSC13,representing 22.94% of the detected core regions[(295+285+267)/3 693]. In addition, these core regions contained 47 515 SNPs, with an average of 12.87 SNPs(ranging from 3 to 20) within each core region.
After filtering, 172 EHH tests and 154 cores remained(Figs. 1 and 2). For all 3 693 core regions, 20 577 EHH tests were performed. The distribution of REHH values vs.haplotype frequencies across the swine genome is shown in Fig. 1. Fig. 2 shows the distribution of REHH values across the whole genome. According the EHH test results,the average haplotype frequency was 0.29.
3.3. Genome annotation
Within the 154 significant core regions, 551 candidate genes were annotated in the NCBI database. A majority of the detected regions were distributed on SSC2, SSC6, and SSC10. The total length of the 154 significant core regions was 16.52 Mb. The top 20 significant core regions are shown in Table 2 and additional details of the 154 significant core regions are shown in Appendix A.
QTL that overlapped with these core regions were found to be associated with important economic traits such as body weight, average daily gain, and teat number. A total of 5 268 QTLs were reported within the significant core regions(Appendix B). The times of reported QTL in each significant core regions are shown in Table 2.

Table 1 Summary of genome-wide markers and core region (CR) distribution in Duroc pigs1)
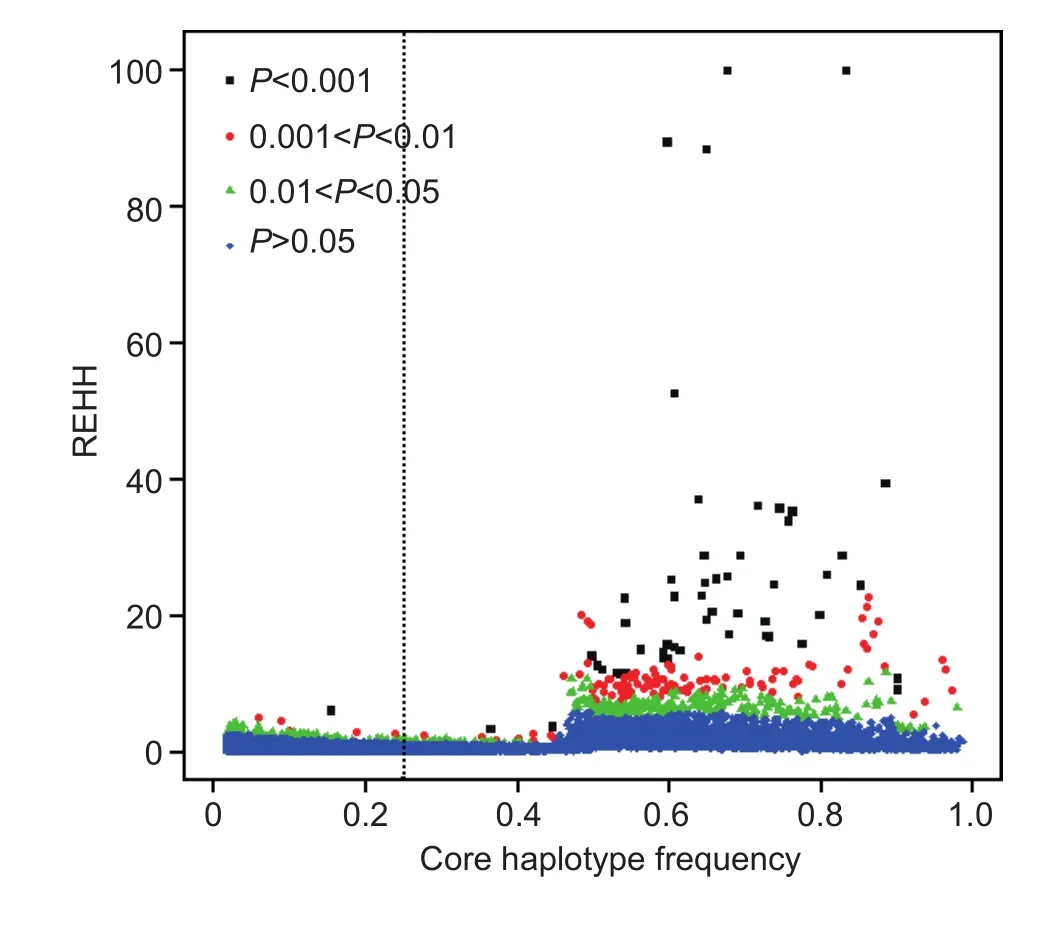
Fig. 1 The distribution of relative extended haplotype homozygosity (REHH) vs. core haplotype frequency in a Duroc pig population. Different P-value ranges are marked by different colors. The vertical dashed line=0.25. Log10REHH was calculated using H=0.1.
3.4. GO terms and KEGG pathway enrichment analysis
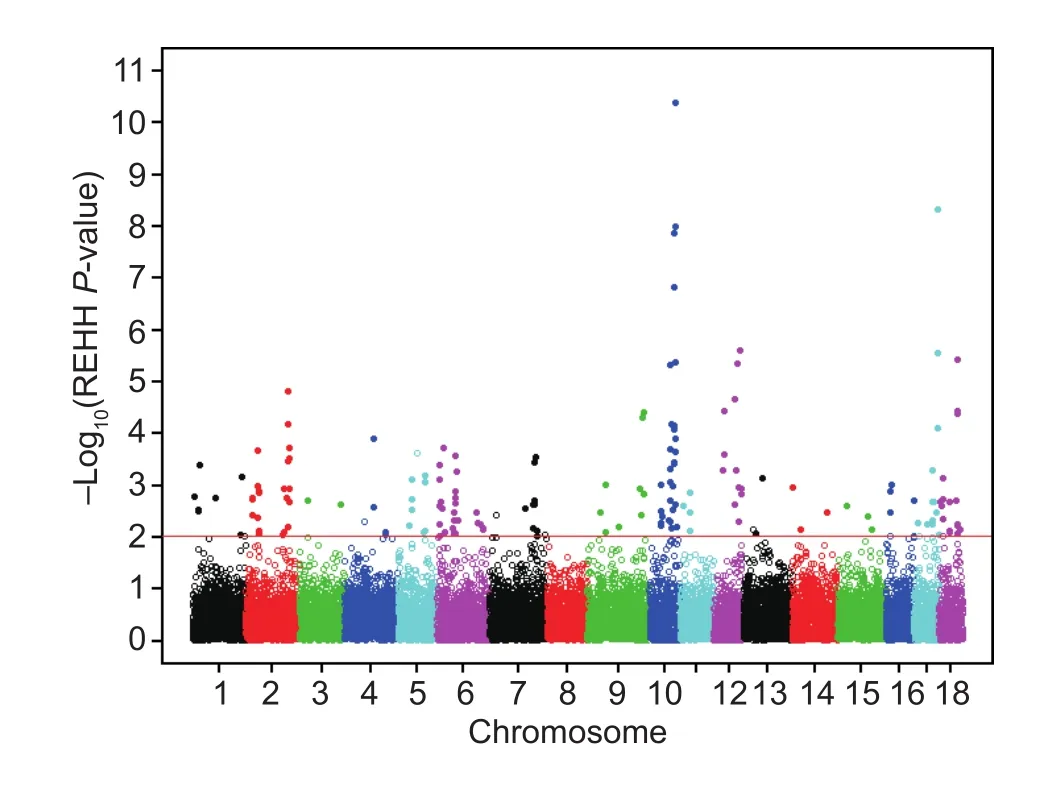
Fig. 2 Manhattan plot with P-values of core haplotypes on the whole genome of the Duroc pig. The hollow data points are cores haplotypes with a frequency>0.25 and REHH P-values<0.01 (-log10(REHH P-value)>2).
Nine KEGG pathways and five GO terms involved 73 candidate genes were targeted (Table 3). The most significant GO term was GO: 0009952, which is defined as anterior/posterior or pattern specification. A total of 15 candidate genes were enriched in the ssc04151:PI3K-Akt signaling pathway, which enriched most of candidate genes.
4. Discussion
The identified significant core regions in this study overlapped with QTLs that were previously associatedwith several economically important traits (e.g., average daily gain, backfat, marbling, teat number, meat quality,and carcass traits). In this study, 154 significant core regions were identified, with overlap between 106 regions under selection (Appendices A and B) and QTL previously reported to be associated with average daily gain.Additional candidate genes were enriched in the GO term GO: 0048704, which is defined as embryonic skeletal system morphogenesis. The QTL mapping and selective signature detection results together suggest that the detected regions in the Duroc genome are reliable.
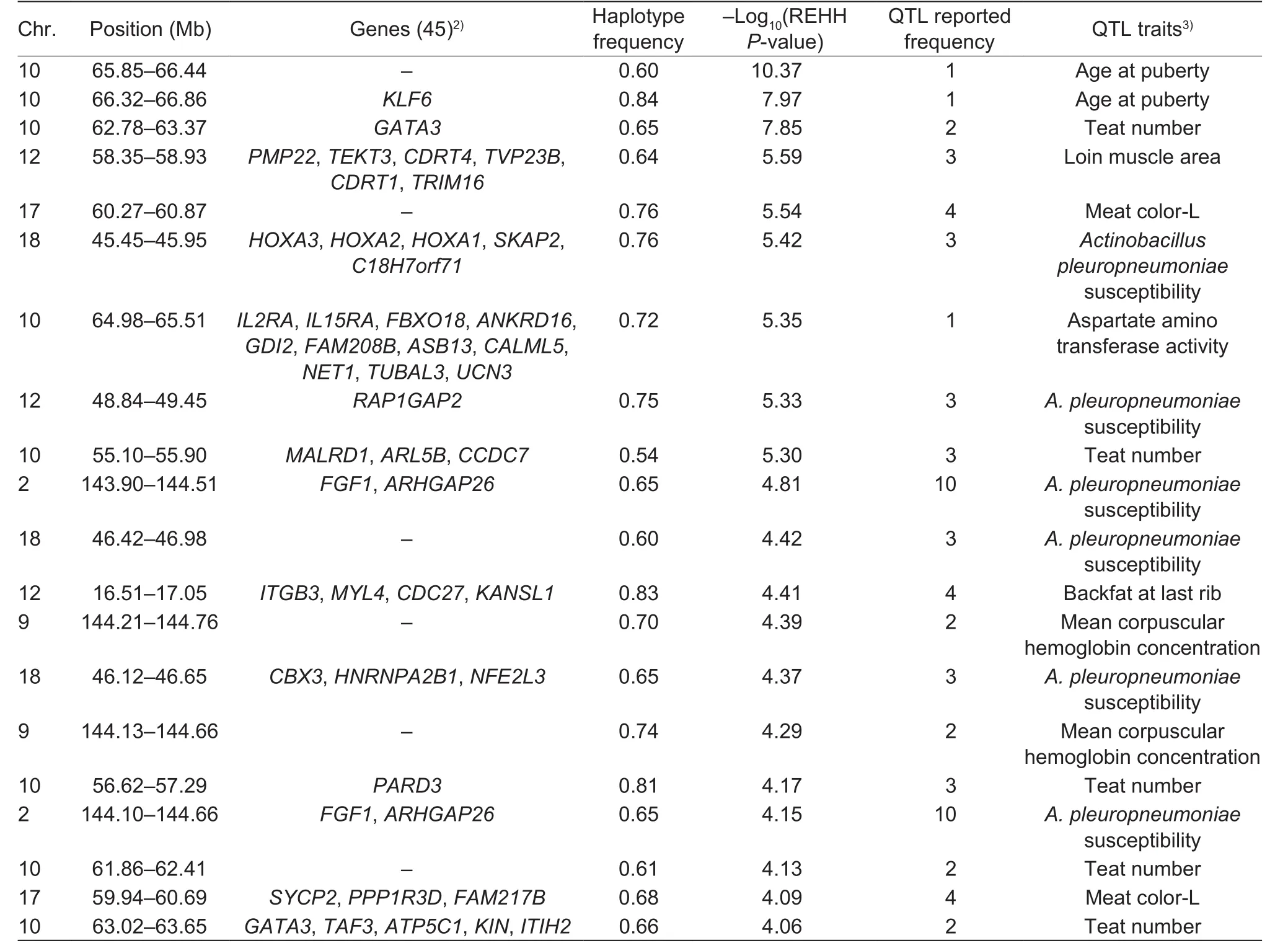
Table 2 Summary of the top 20 significant core genome regions in a Duroc pig population1)
The most significant GO term in the biological process category detected in this study was GO: 0009952, which is defined as anterior/posterior pattern specification(P-value=0.00002). Anterior/posterior pattern specification was previously reported as genes specifying terminal domains (Strecker and Lengyel 1988). The candidate genes involved in this biological process are homeobox, which were first discovered to be conserved between many homeotic genes in Drosophila (Regulski et al. 1985). The proteins containing this DNA binding motif were typically transcription factors (Freund and Mcinnes 1995). These candidate genes can be used to study evolution in wild boars by comparing to bred Duroc populations, and potentially offers new ways to explore evolutionary mechanisms.
As a commercial pig breed, Duroc was once reported for its high intramuscular fat content compared to other widespread commercial pigs (Warriss et al. 1996). In this study, the QTL associated with muscle fat content were found to overlap with five out of 154 significant core regions.Within these 154 significant core regions, QTL associated with muscle tissue were reported 108 times. However,the gene ELOVL3 was not enriched in the significant core regions as reported in Wilkinson et al. (2013).
It is noteworthy that there was one QTL associated with the age at puberty, which overlapped with nine significant core regions. Moreover, age at puberty in Duroc pigs is later than other commercial breeds, like Landrace, Yorkshire(Tummaruk et al. 2014), and Wuzhishan pigs (Min et al.2014). The Duroc pig can serve as a reference population to compare with Chinese indigenous breeds in future studies.
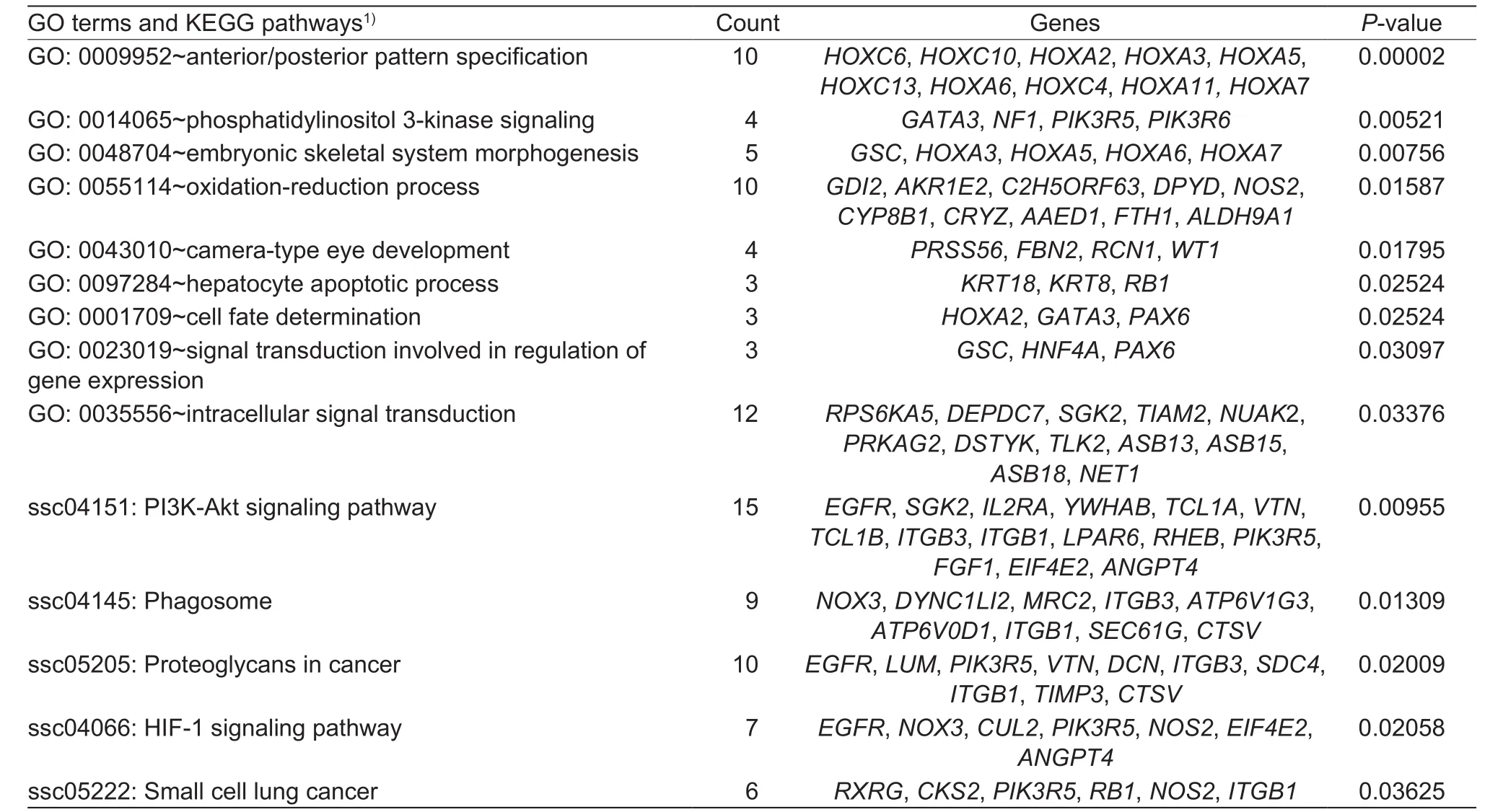
Table 3 GO terms and KEGG pathways enriched with candidate genes
A QTL associated with Actinobacillus pleuropneumoniae susceptibility was reported located in 44 out of the 154 significant core regions, and it may relate to the Duorc origin. These genome regions were not found in other selective signature studies focused on Duroc pigs (Ai et al.2013; Wilkinson et al. 2013; Bosse et al. 2014). Several loci have been proven to be specific in Duroc populations,such as the red coat related gene MC1R (Kijas et al. 1998;Fang et al. 2009), which was not detected in our study.Possible reasons include: (1) the limitation of statistical methods, selection criteria, and pig population size; (2) the pig genome annotation was incomplete at the time of this study; and (3) the low density of the currently available SNP chips. A total of 551 genes reported in a previous study where sequencing data were used to identify selected genes in Duroc population (Choi et al. 2015), were also identified in this study. Therefore, more density marker panels,sequencing data, and larger population sizes are necessary to detect additional genes under selection and to enhance the accuracy of selective signature detection.
5. Conclusion
In this study, 154 significant core regions and some important functional candidate genes were detected in a Duroc pig population. Moreover, these regions overlap with previously reported QTL that are associated with several economically important traits including average daily gain and backfat thickness. Candidate genes annotated within these regions were enriched in several biology pathways such as anterior/posterior pattern specification and the PI3KAkt signaling pathway. This study may provide knowledge for selection mechanisms and breeding practices in Duroc and other pigs.
Acknowledgements
This research was supported by the earmarked fund for the China Agriculture Research System (CARS-35), the National Natural Science Foundation of China (31772556),the Basic Work of Science and Technology Project, China(2014FY120800), the Pearl River S&T Nova Program of Guangzhou, China (201506010027), and the Guangdong S&T Project, China (2017A020208043).
Appendicesassociated with this paper can be available on http://www.ChinaAgriSci.com/V2/En/appendix.htm
 Journal of Integrative Agriculture2018年11期
Journal of Integrative Agriculture2018年11期
- Journal of Integrative Agriculture的其它文章
- First report of Athelia bombacina causing postharvest fruit rot on pear
- Sustainability assessment of potato fields using the DEXi decision support system in Hamadan Province, lran
- Effect of long-term continuous cropping of strawberry on soil bacterial community structure and diversity
- Alternate row mulching optimizes soil temperature and water conditions and improves wheat yield in dryland farming
- lnter-annual changes in the aggregate-size distribution and associated carbon of soil and their effects on the straw-derived carbon incorporation under long-term no-tillage
- Mycoplasma leachii causes polyarthritis in calves via the blood route but is not associated with pneumonia